

Three-dimensional genome landscape of primary human cancers
- PMID: 40355593
- PMCID: PMC12081301
- DOI: 10.1038/s41588-025-02188-0
Three-dimensional genome landscape of primary human cancers
Abstract
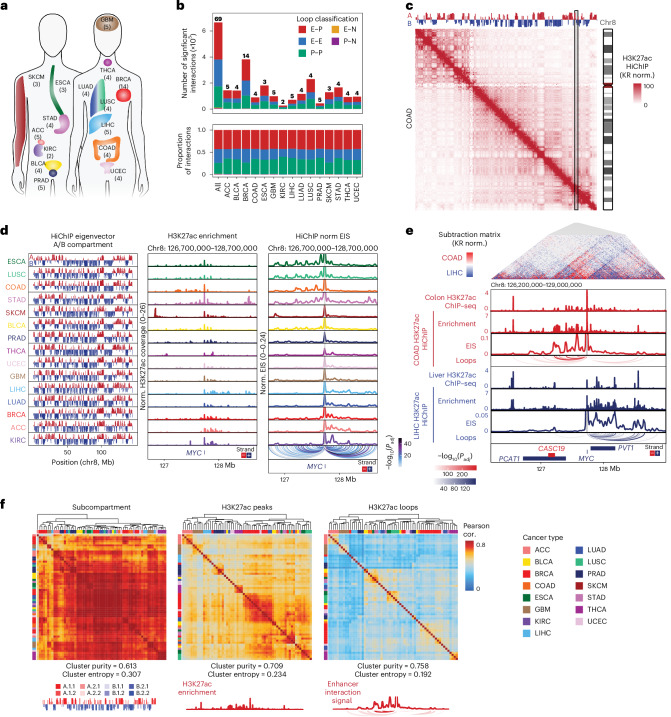
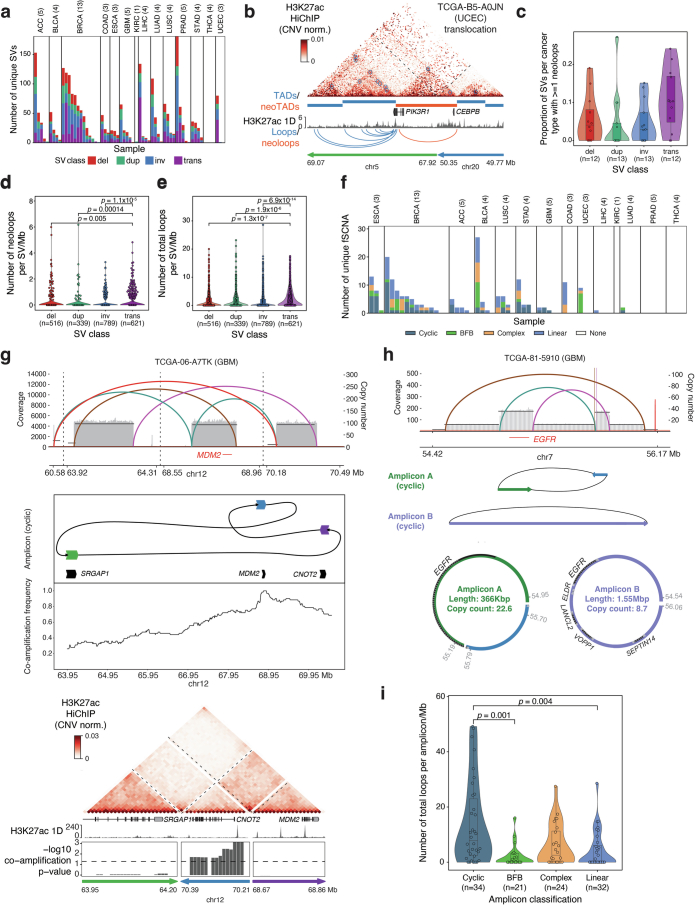
Genome conformation underlies transcriptional regulation by distal enhancers, and genomic rearrangements in cancer can alter critical regulatory interactions. Here we profiled the three-dimensional genome architecture and enhancer connectome of 69 tumor samples spanning 15 primary human cancer types from The Cancer Genome Atlas. We discovered the following three archetypes of enhancer usage for over 100 oncogenes across human cancers: static, selective gain or dynamic rewiring. Integrative analyses revealed the enhancer landscape of noncancer cells in the tumor microenvironment for genes related to immune escape. Deep whole-genome sequencing and enhancer connectome mapping provided accurate detection and validation of diverse structural variants across cancer genomes and revealed distinct enhancer rewiring consequences from noncoding point mutations, genomic inversions, translocations and focal amplifications. Extrachromosomal DNA promoted more extensive enhancer rewiring among several types of focal amplification mechanisms. These results suggest a systematic approach to understanding genome topology in cancer etiology and therapy.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: H.Y.C. is a cofounder of Accent Therapeutics, Boundless Bio, Cartography Biosciences and Orbital Therapeutics; was an advisor of 10x Genomics, Arsenal Biosciences, Chroma Medicine and Spring Discovery until 15 December 2024 and is an employee and stockholder of Amgen as of 16 December 2024. K.E.Y. is a consultant for Cartography Biosciences. A.D.C. receives research funding from Bayer and is a consultant for KaryoVerse. P.W.L. is an advisor for Tagomics, FOXO Biosciences and AnchorDX. W.J.G. is named as an inventor on patents describing ATAC–seq methods. 10x Genomics has licensed intellectual property on which W.J.G. is listed as an inventor. W.J.G. holds options in 10x Genomics and is a consultant for Ultima Genomics and Guardant Health. W.J.G. is a scientific cofounder of Protillion Biosciences. V.B. is a cofounder, serves on the scientific advisory board of Boundless Bio and Abterra and holds equity in both companies. The other authors declare no competing interests.
Figures












References
MeSH terms
Grants and funding
- DP1HG013599/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- 1U24CA264023-01/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- F99 CA274692/CA/NCI NIH HHS/United States
- F99 CA253729/CA/NCI NIH HHS/United States
- U24 CA264029/CA/NCI NIH HHS/United States
- CGCATF-2021/100025/Cancer Research UK (CRUK)
- R01GM114362/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01 CA218668/CA/NCI NIH HHS/United States
- U24CA264379/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- 1U24CA264029-01/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01NS128028/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- U24 CA264009/CA/NCI NIH HHS/United States
- R01CA218668/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- RM1 HG007735/HG/NHGRI NIH HHS/United States
- NIH F99CA274692/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- DP1 HG013599/HG/NHGRI NIH HHS/United States
- R01 HL171611/HL/NHLBI NIH HHS/United States
- U24 CA264379/CA/NCI NIH HHS/United States
- U24 CA264032/CA/NCI NIH HHS/United States
- DGE-1656518/National Science Foundation (NSF)
- U24 CA264021/CA/NCI NIH HHS/United States
- RM1-HG007735/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- R01 NS136475/NS/NINDS NIH HHS/United States
- R35-CA209919/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- OT2 CA278635/CA/NCI NIH HHS/United States
- R01 NS128028/NS/NINDS NIH HHS/United States
- U24CA264032/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- OT2CA278635/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- CGCSDF-2021\100007/Cancer Research UK (CRUK)
- U24 CA264023/CA/NCI NIH HHS/United States
- R01 GM114362/GM/NIGMS NIH HHS/United States
- R35 CA209919/CA/NCI NIH HHS/United States
- NIH F99CA253729/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R01HL171611/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
LinkOut - more resources
Full Text Sources
Medical
