

Development of a machine learning-based predictive risk model combining fatty acid metabolism and ferroptosis for immunotherapy response and prognosis in prostate cancer
- PMID: 40355680
- PMCID: PMC12069205
- DOI: 10.1007/s12672-025-02484-5
Development of a machine learning-based predictive risk model combining fatty acid metabolism and ferroptosis for immunotherapy response and prognosis in prostate cancer
Abstract
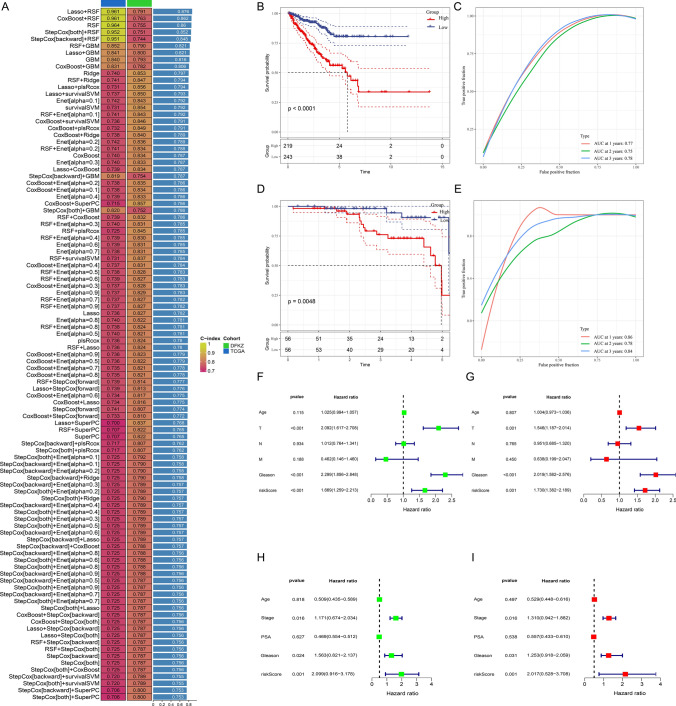
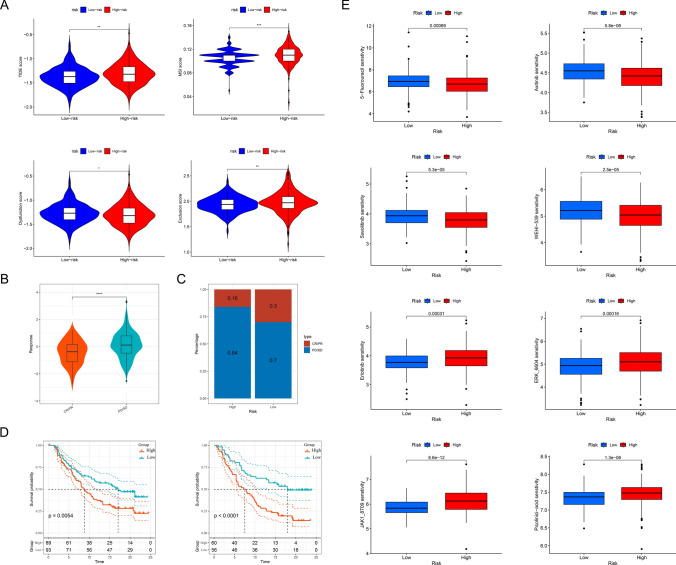
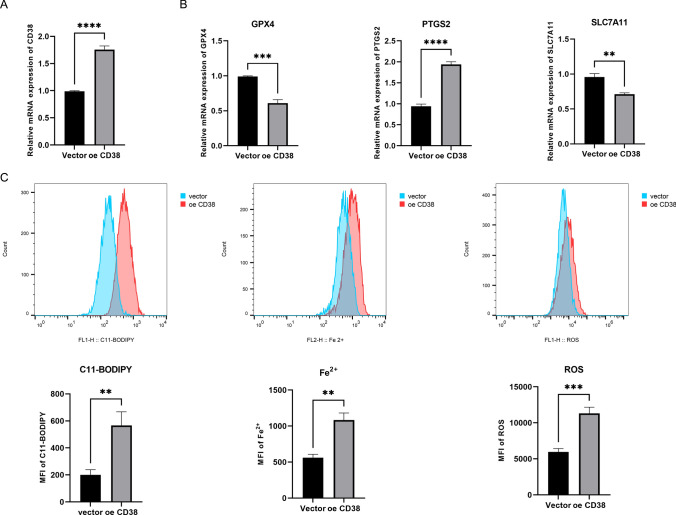
Prostate cancer (PCa) remains a leading cause of cancer-related mortality, necessitating robust prognostic models and personalized therapeutic strategies. This study integrated bulk RNA sequencing, single-cell RNA sequencing (scRNA-seq), and spatial transcriptomics to construct a prognostic model based on genes shared between ferroptosis and fatty acid metabolism (FAM). Using the TCGA-PRAD dataset, we identified 73 differentially expressed genes (DEGs) at the intersection of ferroptosis and FAM, of which 19 were significantly associated with progression-free survival (PFS). A machine learning-based prognostic model, optimized using the Lasso + Random Survival Forest (RSF) algorithm, achieved a high C-index of 0.876 and demonstrated strong predictive accuracy (1-, 2-, and 3-year AUCs: 0.77, 0.75, and 0.78, respectively). The model, validated in the DFKZ cohort, stratified patients into high- and low-risk groups, with the high-risk group exhibiting worse PFS and higher tumor mutation burden (TMB). Functional enrichment analysis revealed distinct pathway activities, with high-risk patients showing enrichment in immune-related and proliferative pathways, while low-risk patients were enriched in metabolic pathways. Immune microenvironment analysis revealed heightened immune activity in high-risk patients, characterized by increased infiltration of CD8 + T cells, regulatory T cells, and M2 macrophages, alongside elevated TIDE scores, suggesting immune evasion and resistance to immunotherapy. In contrast, low-risk patients exhibited higher infiltration of plasma cells and neutrophils and demonstrated better responses to immune checkpoint inhibitors (ICIs). Spatial transcriptomics and scRNA-seq further elucidated the spatial distribution of model genes, highlighting the central role of macrophages in mediating risk stratification. Additionally, chemotherapy sensitivity analysis identified potential therapeutic agents, such as Erlotinib and Picolinic acid, for low-risk patients. In vitro experiments showed that overexpression of CD38 in the PC-3 cell line led to elevated lipid peroxidation (C11-BODIPY) and reactive oxygen species (ROS), suggesting increased cell ferroptosis. These findings provide a comprehensive framework for risk stratification and personalized treatment in PCa, bridging molecular mechanisms with clinical outcomes.
Keywords: Fatty acid metabolism; Ferroptosis; Machine learning; Multi-omics; Prostate cancer; Tumor microenvironment.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not available, for the study used only public database samples and did not involve collecting human or animal specimens for experiments. Competing interests: The authors declare no competing interests.
Figures







References
Grants and funding
- 202201020582/Science and Technology Planning Project of Guangzhou City
- LJKZZ20220100/the Scientific Research Project of Ministry of Education of Liaoning Province
- JCHZ2023001/the Interdisciplinary Research Cooperation Project Team Funding of Dalian Medical University Planning and research category (focusing on planning for recreation)
- DMU-2&DICP UN202304/the United Foundation for Dalian Institute of Chemical Physics & Chinese Academy of Sciences and the Second Hospital of DaliarMedical University
- 2022LCJSGC16/"1+X "program for Clinical Competency enhancement-Clinical Technical Level Improvement Project, The Second Hospital of Dalian Medical University
LinkOut - more resources
Full Text Sources
Research Materials