

Metabolic mapping of the human solute carrier superfamily
- PMID: 40355754
- PMCID: PMC12130231
- DOI: 10.1038/s44320-025-00106-4
Metabolic mapping of the human solute carrier superfamily
Abstract
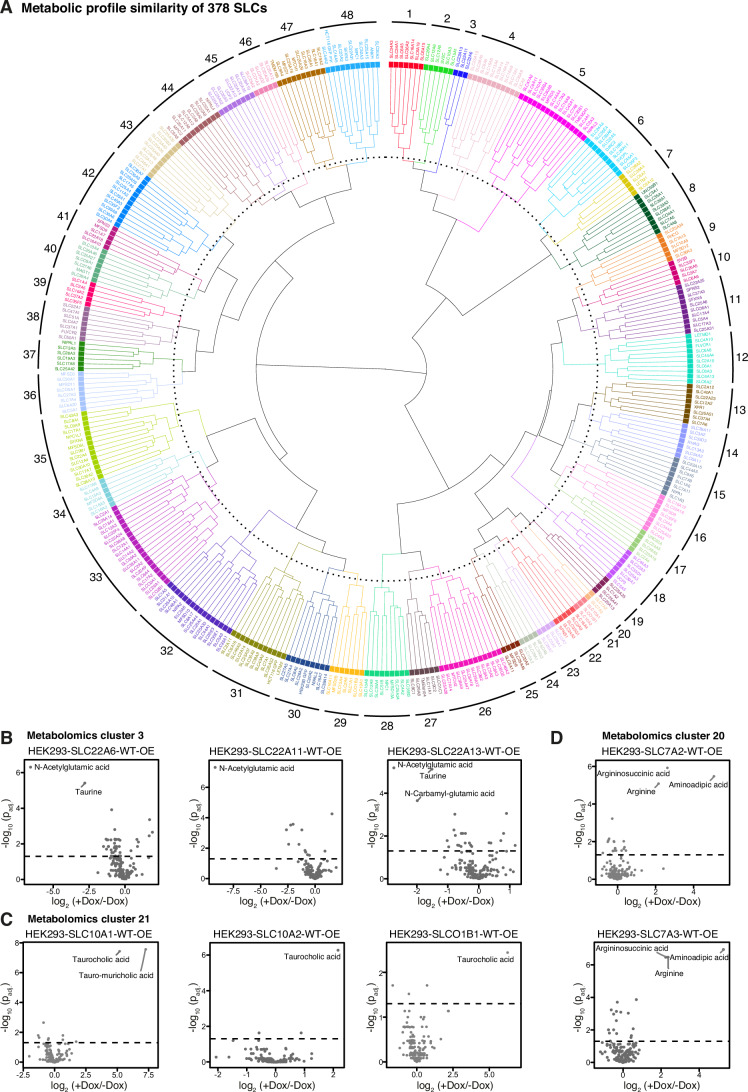
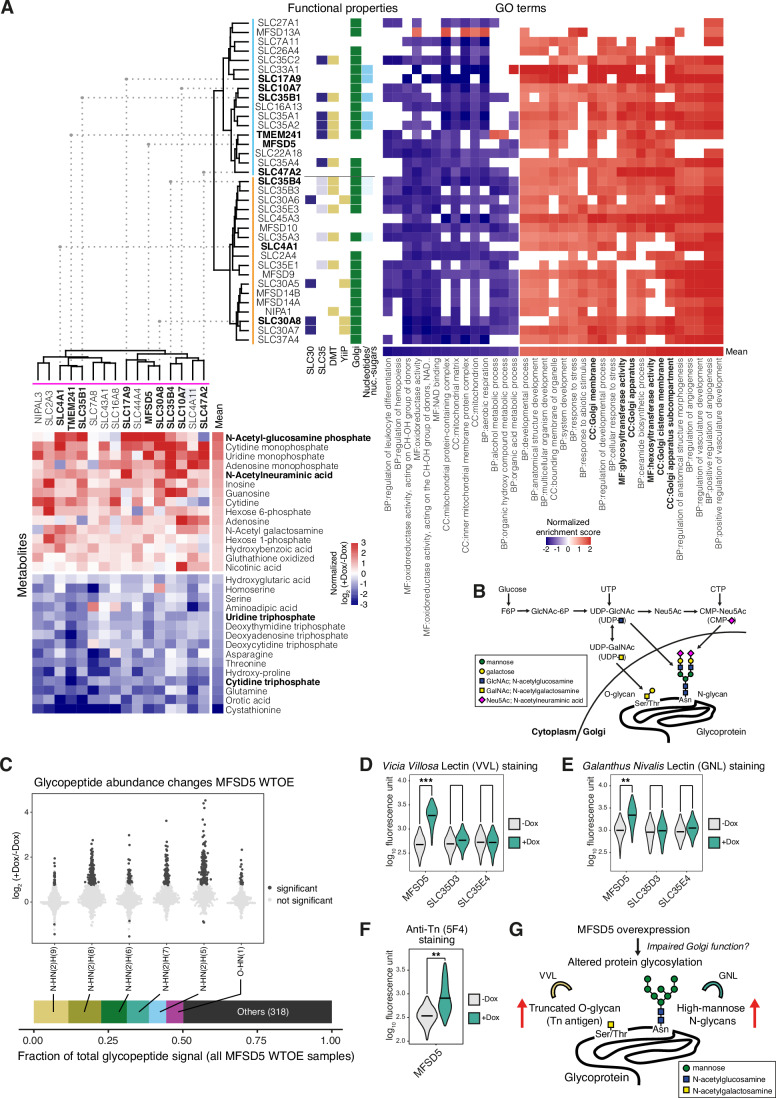
Solute carrier (SLC) transporters govern most of the chemical exchange across cellular membranes and are integral to metabolic regulation, which in turn is linked to cellular function and identity. Despite their key role, individual functions of the SLC superfamily members were not evaluated systematically. We determined the metabolic and transcriptional profiles upon SLC overexpression in knock-out or wild-type isogenic cell backgrounds for 378 SLCs and 441 SLCs, respectively. Targeted metabolomics provided a fingerprint of 189 intracellular metabolites, while transcriptomics offered insights into cellular programs modulated by SLC expression. Beyond the metabolic profiles of 102 SLCs directly related to their known substrates, we identified putative substrates or metabolic pathway connections for 71 SLCs without previously annotated bona fide substrates, including SLC45A4 as a new polyamine transporter. By comparing the molecular profiles, we identified functionally related SLC groups, including some with distinct impacts on osmolyte balancing and glycosylation. The assessment of functionally related human genes presented here may serve as a blueprint for other systematic studies and supports future investigations into the functional roles of SLCs.
Keywords: Membrane Transporters; Metabolism; Metabolomics; Solute Carriers; Transcriptomics.
© 2025. The Author(s).
Conflict of interest statement
Disclosure and competing interests statement. GS-F is a co-founder and owns shares of Solgate GmbH, an SLC-focused company.
Figures









References
-
- Agrawal S, Kumar S, Sehgal R, George S, Gupta R, Poddar S, Jha A, Pathak S (2019) El-MAVEN: a fast, robust, and user-friendly mass spectrometry data processing engine for metabolomics. Methods Mol Biol 1978:301–321 - PubMed
-
- Azzollini L, Prete DD, Wolf G, Klimek C, Saggioro M, Ricci F, Christodoulaki E, Wiedmer T, Ingles-Prieto A, Superti-Furga G et al (2024) Development of a live cell assay for the zinc transporter ZnT8. SLAS Discov 29:100166 - PubMed
-
- Baker SA, Rutter J (2023) Metabolites as signalling molecules. Nat Rev Mol Cell Biol 24:355–374 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
