

ATIP1 Is a Suppressor of Cardiac Hypertrophy and Modulates AT2-Dependent Signaling in Cardiac Myocytes
- PMID: 40358169
- PMCID: PMC12071853
- DOI: 10.3390/cells14090645
ATIP1 Is a Suppressor of Cardiac Hypertrophy and Modulates AT2-Dependent Signaling in Cardiac Myocytes
Abstract
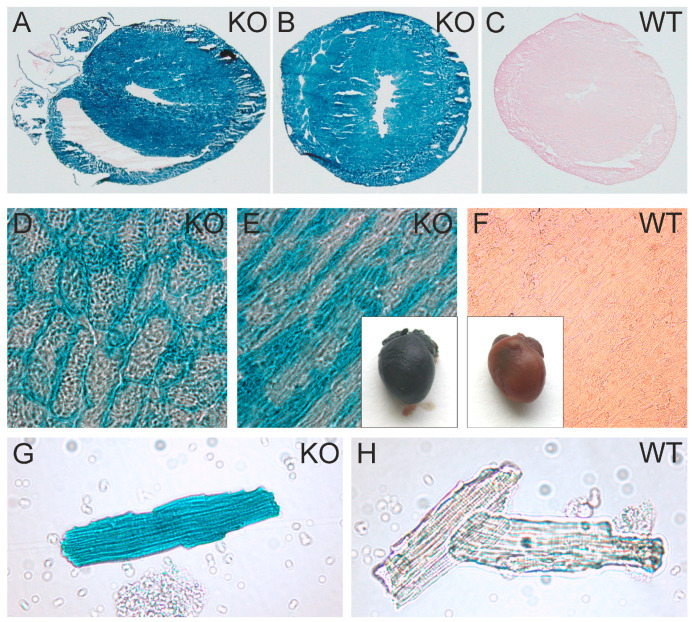
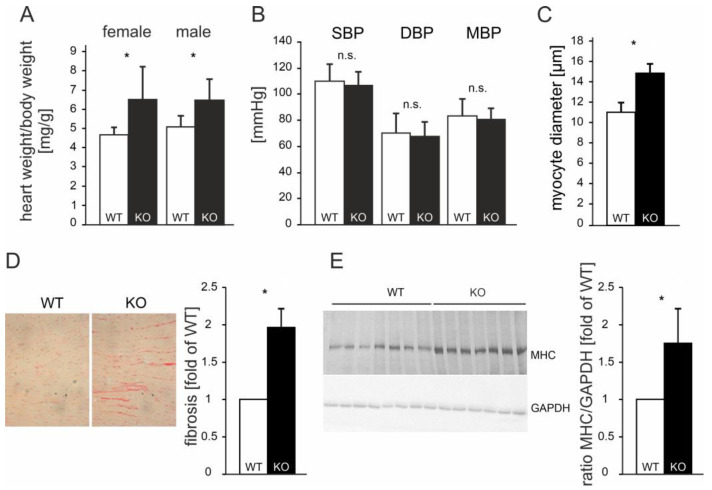
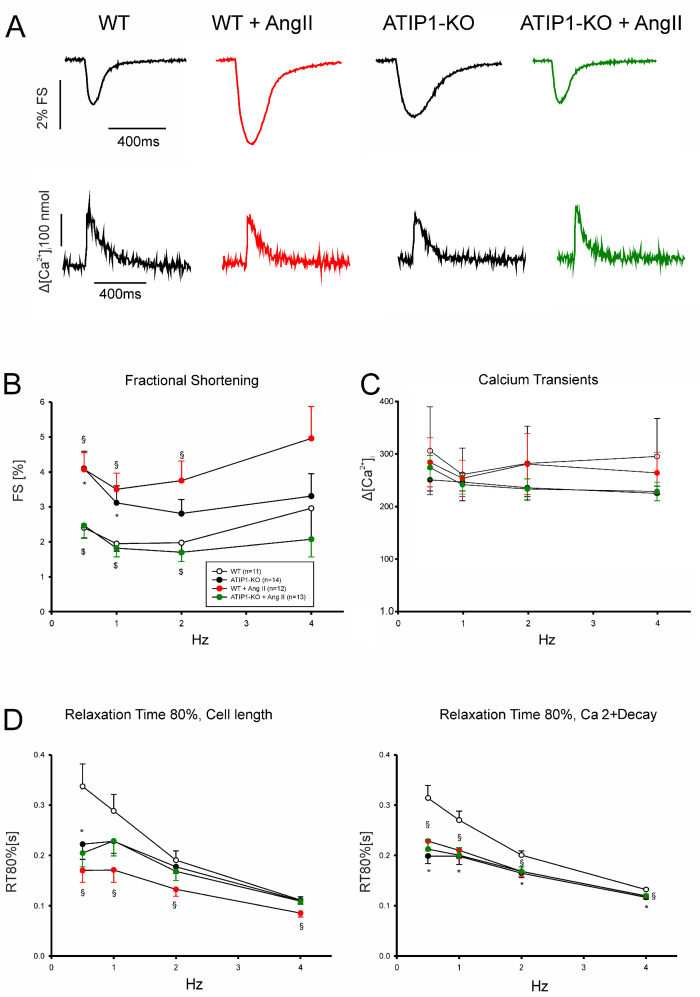
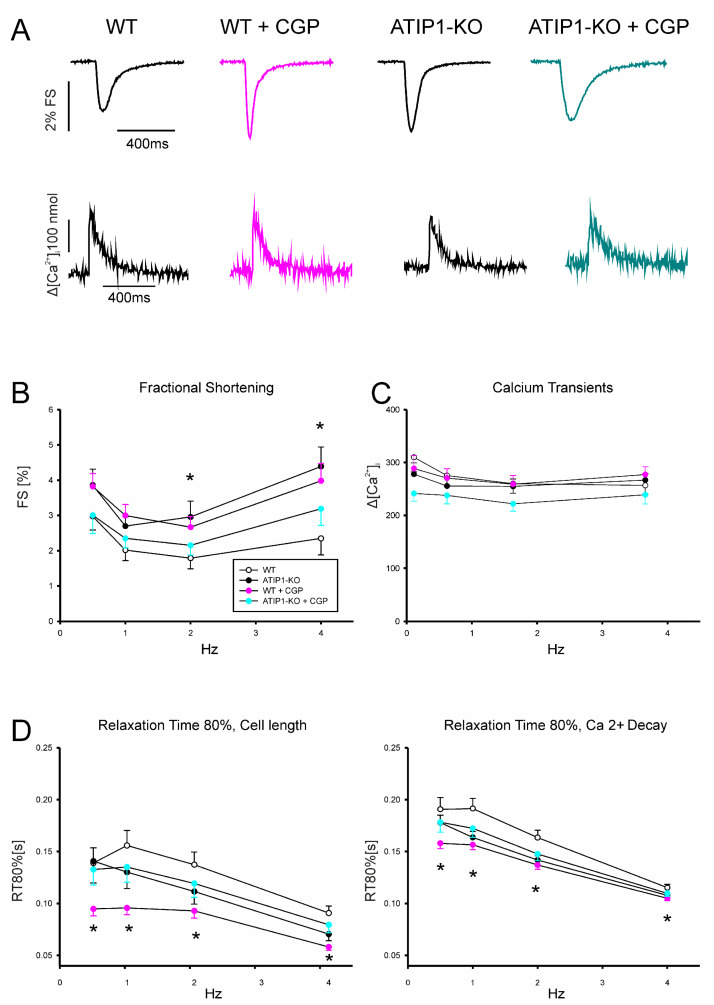
So far, the molecular functions of the angiotensin-type-2 receptor (AT2) interacting protein (ATIP1) have remained unclear, although expression studies have revealed high levels of ATIP1 in the heart. To unravel its physiological function, we investigated ATIP1-KO mice. They develop a spontaneous cardiac hypertrophy with a significantly increased heart/bodyweight ratio, enlarged cardiomyocyte diameters, and augmented myocardial fibrosis. Hemodynamic measurements revealed an increased ejection fraction (EF) in untreated ATIP1-KO mice, and reduced end-systolic and end-diastolic volumes (ESV and EDV), which, in sum, reflect a compensated concentric cardiac hypertrophy. Importantly, no significant differences in blood pressure (BP) were observed. Chronic angiotensin II (AngII) infusion resulted in increases in BP and EF in ATIP1-KO and WT mice. Reductions in ESV and EDV occurred in both ATIP1-KO and WT but to a lesser extent in ATIP1-KOs. Isolated cardiomyocytes exhibited a significantly increased contractility in ATIP1-KO and accelerated Ca2+ decay. AngII treatment resulted in increased fractional shortening in WT but decreased shortening in ATIP1-KO, accompanied by accelerated cell relaxation in WT but absent effects on relaxation in ATIP1-KO cells. The AT2 agonist CGP42112A increased shortening in WT cardiomyocytes but, again, did not affect shortening in ATIP1-KO cells. Relaxation was accelerated by CGP42112A in WT but was unaffected in ATIP1-KO cells. We show that ATIP1 deficiency results in spontaneous cardiac hypertrophy in vivo and that ATIP1 is a downstream signal in the AT2 pathway regulating cell contractility. We hypothesize that the latter effect is because of a disinhibition of the AT1 pathway by impaired AT2 signaling.
Keywords: ATIP1; MTUS1; angiotensin-type-2 receptor; cardiac hypertrophy; knockout mice.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures

Similar articles
-
Impact of AT2 receptor deficiency on postnatal cardiovascular development.PLoS One. 2012;7(10):e47916. doi: 10.1371/journal.pone.0047916. Epub 2012 Oct 29. PLoS One. 2012. PMID: 23144713 Free PMC article.
-
The angiotensin II type 2 receptor causes constitutive growth of cardiomyocytes and does not antagonize angiotensin II type 1 receptor-mediated hypertrophy.Hypertension. 2005 Dec;46(6):1347-54. doi: 10.1161/01.HYP.0000193504.51489.cf. Epub 2005 Nov 14. Hypertension. 2005. PMID: 16286564
-
Promyelocytic leukemia zinc finger protein activates GATA4 transcription and mediates cardiac hypertrophic signaling from angiotensin II receptor 2.PLoS One. 2012;7(4):e35632. doi: 10.1371/journal.pone.0035632. Epub 2012 Apr 27. PLoS One. 2012. PMID: 22558183 Free PMC article.
-
Ablation of lncRNA Miat attenuates pathological hypertrophy and heart failure.Theranostics. 2021 Jul 6;11(16):7995-8007. doi: 10.7150/thno.50990. eCollection 2021. Theranostics. 2021. PMID: 34335976 Free PMC article.
-
The renin-angiotensin system and experimental heart failure.Cardiovasc Res. 1999 Sep;43(4):838-49. doi: 10.1016/s0008-6363(99)00145-5. Cardiovasc Res. 1999. PMID: 10615411 Review.
References
-
- de Gasparo M., Catt K.J., Inagami T., Wright J.W., Unger T. International union of pharmacology. Xxiii. The angiotensin ii receptors. Pharmacol. Rev. 2000;52:415–472. - PubMed
-
- Lindberg H., Nielsen D., Jensen B.V., Eriksen J., Skovsgaard T. Angiotensin converting enzyme inhibitors for cancer treatment? Acta Oncol. 2004;43:142–152. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
