

Acute coronary syndromes: mechanisms, challenges, and new opportunities
- PMID: 40358623
- PMCID: PMC12314746
- DOI: 10.1093/eurheartj/ehaf289
Acute coronary syndromes: mechanisms, challenges, and new opportunities
Abstract
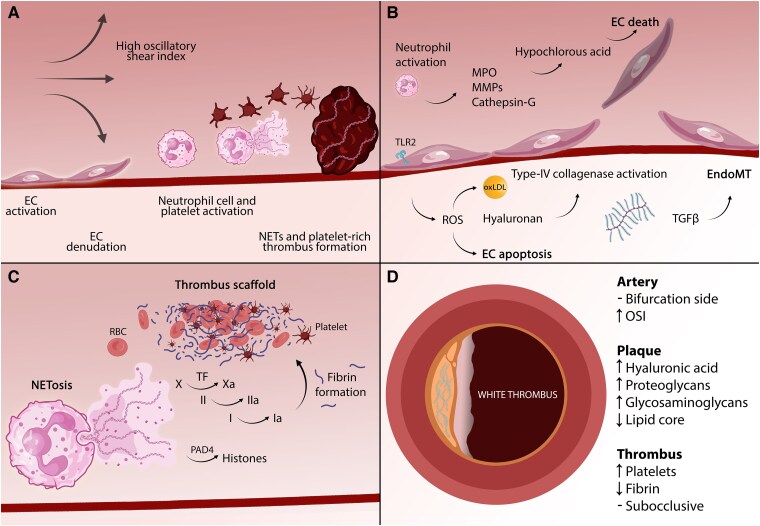
Despite advances in research and patient management, atherosclerosis and its dreaded acute and chronic sequelae continue to account for one out of three deaths globally. The vast majority of acute coronary syndromes (ACS) arise from either plaque rupture or erosion, but other mechanisms, including calcific nodules, embolism, spontaneous coronary artery dissection, coronary spasm, and microvascular dysfunction, can also cause ACS. This ACS heterogeneity necessitates a paradigm shift in its management that extends beyond the binary interpretation of electrocardiographic and biomarker data. Indeed, given the evolution in the global risk factor profile, the increasing importance of previously underappreciated mechanisms, the evolving appreciation of sex-specific disease characteristics, and the advent of rapidly evolving technologies, a precision medicine approach is warranted. This review provides an update of the mechanisms of ACS, delineates the role of previously underappreciated contributors, discusses sex-specific differences, and explores novel tools for contemporary and personalized management of patients with ACS. Beyond mechanistic insights, it examines evolving imaging techniques, biomarkers, and regression- and machine learning-based approaches for the diagnosis (e.g. CoDE-ACS, MI3) and prognosis (e.g. PRAISE, GRACE, SEX-SHOCK scores) of ACS, along with their implications for future ACS management. A more individualized approach to patients with ACS is advocated, emphasizing the need for innovative studies on emerging technologies, including artificial intelligence, which may collectively facilitate clinical decision-making within a more mechanistic framework, thereby personalizing patient care and potentially improving long-term outcomes.
Keywords: Acute coronary syndromes; Acute myocardial infarction; Artificial intelligence; C-reactive protein; Clinical decision-making; Dual antiplatelet therapy; Individualized management; Inflammation; Intravascular ultrasound; Machine learning; Optical coherence tomography; Percutaneous coronary intervention; Plaque erosion; Plaque healing; Plaque rupture; Precision medicine; Sex differences.
© The Author(s) 2025. Published by Oxford University Press on behalf of the European Society of Cardiology.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
