

Nanopore direct RNA sequencing of human transcriptomes reveals the complexity of mRNA modifications and crosstalk between regulatory features
- PMID: 40359935
- PMCID: PMC12230237
- DOI: 10.1016/j.xgen.2025.100872
Nanopore direct RNA sequencing of human transcriptomes reveals the complexity of mRNA modifications and crosstalk between regulatory features
Abstract
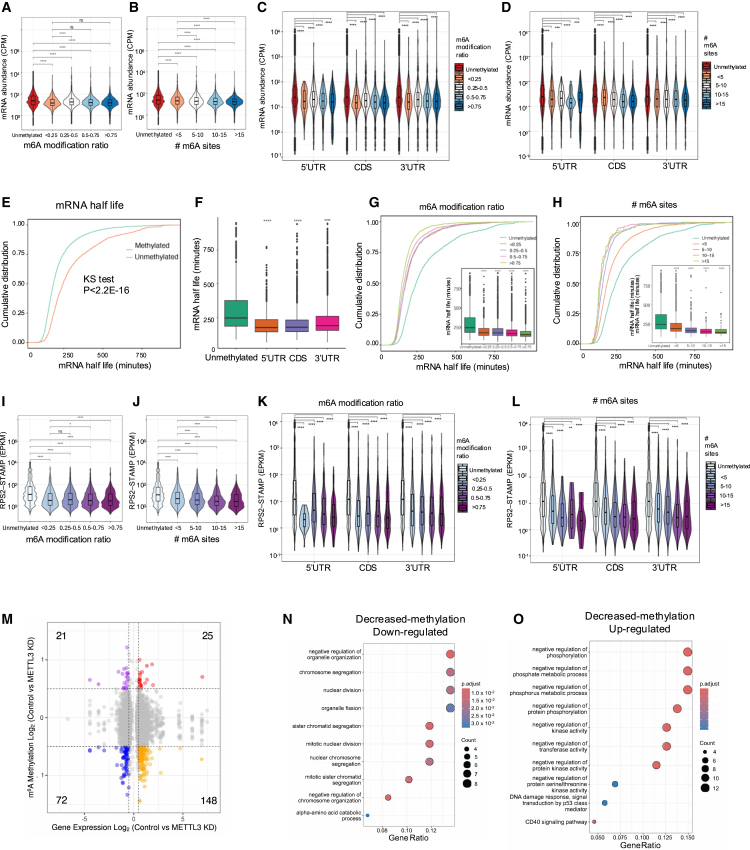
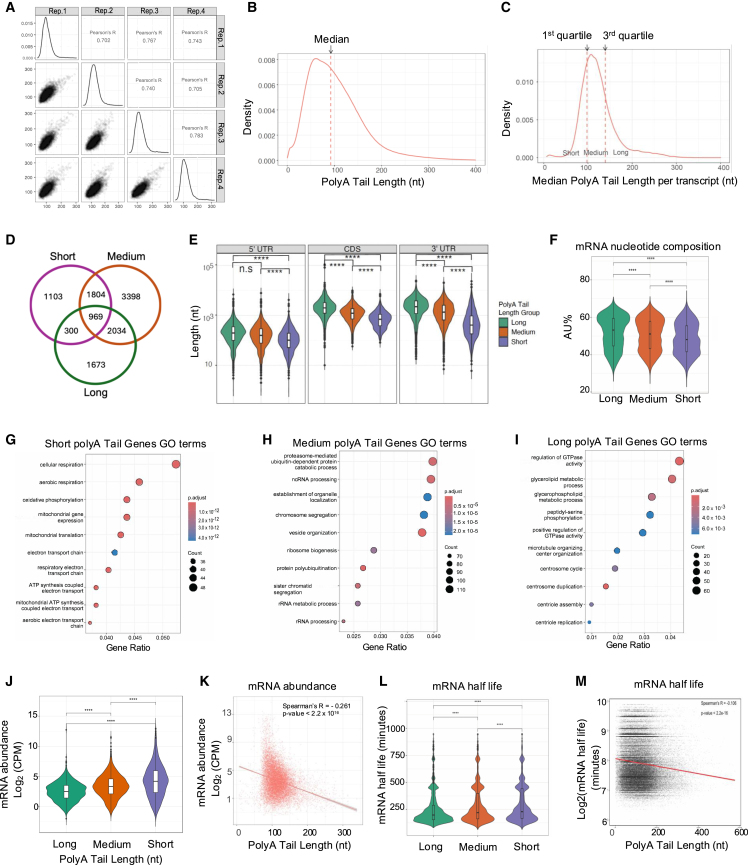
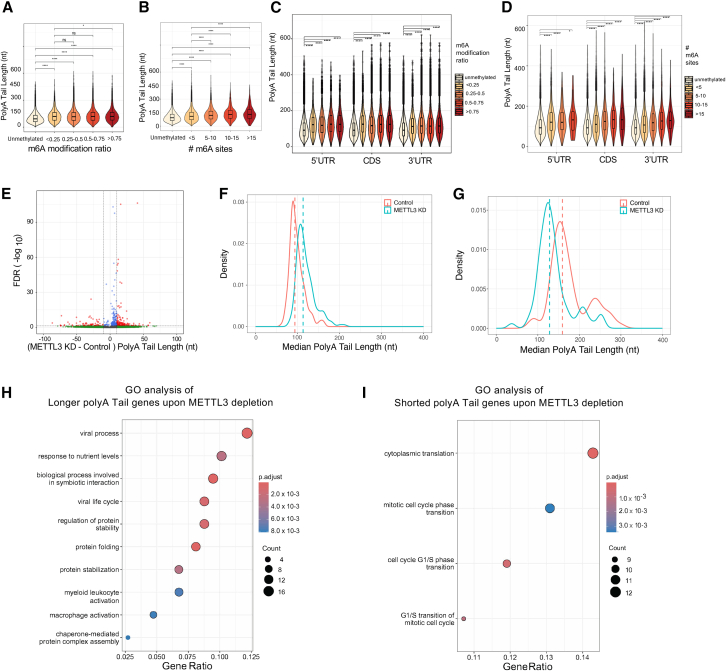
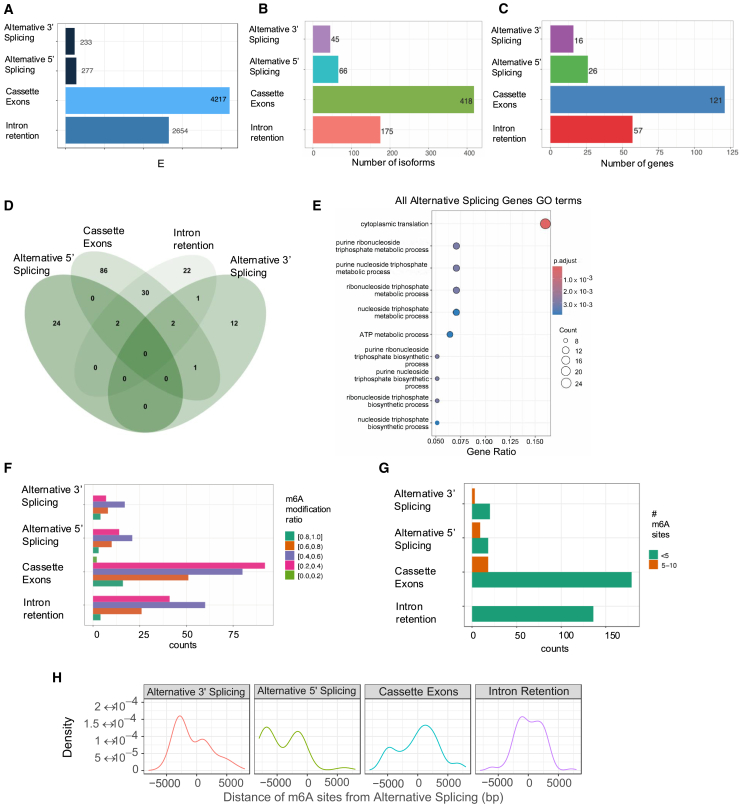
The identification and functional characterization of chemical modifications on an mRNA molecule, in particular N6-methyladenosine (m6A) modification, significantly broadened our understanding of RNA function and regulation. While interactions between RNA modifications and other RNA features have been proposed, direct evidence showing correlation is limited. Here, using Oxford Nanopore long-read direct RNA sequencing (dRNA-seq), we simultaneously interrogate the transcriptome and epitranscriptome of a human leukemia cell line to investigate the correlation between m6A modifications, mRNA abundance, mRNA stability, polyadenylation (poly(A)) tail length, and alternative splicing. High-quality dRNA-seq is important for unbiased and large-scale correlative analyses. Global assessments indicated a negative association between poly(A) tail length and mRNA abundance while uncovering pathway-specific responses upon depletion of the m6A-forming enzyme METTL3. Overall, our study presented a rich dRNA-seq data resource that has been validated and can be further exploited to inquire into the complexity of RNA modifications and potential interplays between RNA regulatory elements.
Keywords: RNA features; RNA methylation; RNA processing; RNA splicing; epitranscriptomics; long-read sequencing; mRNA decay; nanopore direct RNA sequencing; native RNA sequencing; poly(A) tail length.
Copyright © 2025 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors have no competing interests to declare.
Figures


References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
