

Reannotation of cancer mutations based on expressed RNA transcripts reveals functional non-coding mutations in melanoma
- PMID: 40359938
- PMCID: PMC12256795
- DOI: 10.1016/j.ajhg.2025.04.005
Reannotation of cancer mutations based on expressed RNA transcripts reveals functional non-coding mutations in melanoma
Abstract
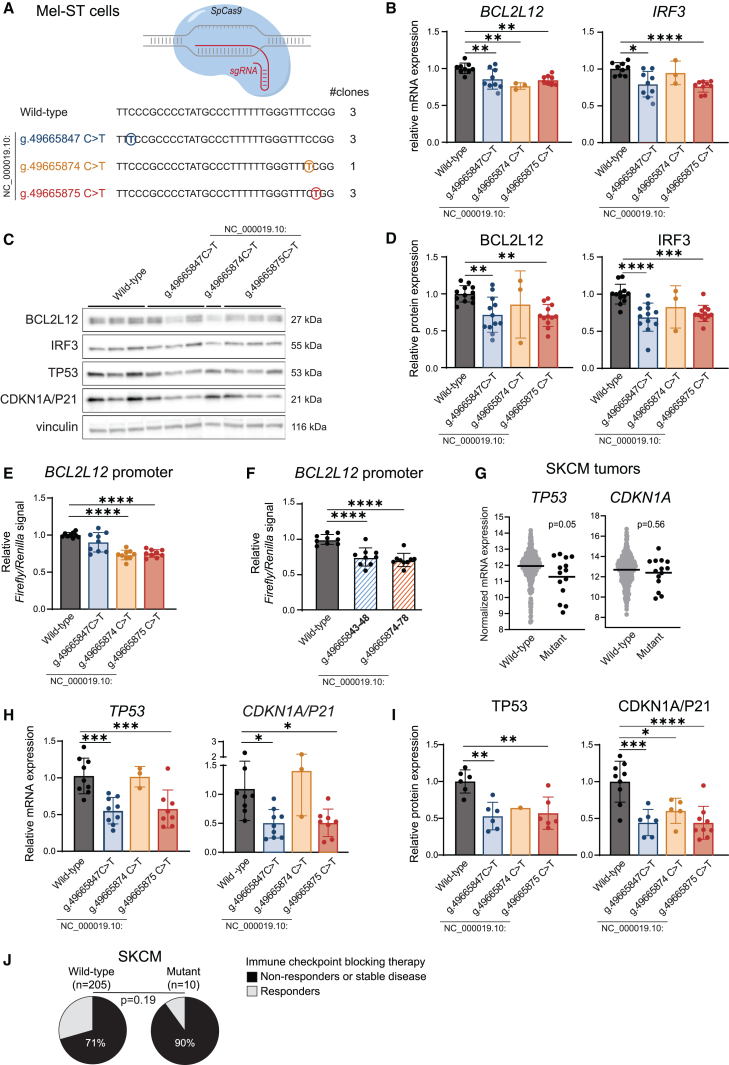
The role of synonymous mutations in cancer pathogenesis is currently underexplored. We developed a method to detect significant clusters of synonymous and missense mutations in public cancer genomics data. In melanoma, we show that 22% (11/50) of these mutation clusters are misannotated as coding mutations because the reference transcripts used for their annotation are not expressed. Instead, these mutations are actually non-coding. This, for instance, applies to the mutation clusters targeting known cancer genes kinetochore localized astrin (SPAG5) binding protein (KNSTRN) and BCL2-like 12 (BCL2L12), each affecting 4%-5% of melanoma tumors. For the latter, we show that these mutations are functional non-coding mutations that target the shared promoter region of interferon regulatory factor 3 (IRF3) and BCL2L12. This results in downregulation of IRF3, BCL2L12, and tumor protein p53 (TP53) expression in a CRISPR-Cas9 primary melanocyte model and in melanoma tumors. In individuals with melanoma, these mutations were also associated with a worse response to immunotherapy. Finally, we propose a simple automated method to more accurately annotate cancer mutations based on expressed transcripts. This work shows the importance of integrating DNA- and RNA-sequencing data to properly annotate mutations and identifies a number of previously overlooked and wrongly annotated functional non-coding mutations in melanoma.
Keywords: CRISPR-Cas9; bioinformatics; expressed transcript; functional genomics; gene regulation; melanoma; mutation annotation; non-coding mutations; synonymous mutations.
Copyright © 2025 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no conflicts of interest.
Figures






References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
