

Extensively acquired antimicrobial-resistant bacteria restructure the individual microbial community in post-antibiotic conditions
- PMID: 40360555
- PMCID: PMC12075632
- DOI: 10.1038/s41522-025-00705-x
Extensively acquired antimicrobial-resistant bacteria restructure the individual microbial community in post-antibiotic conditions
Abstract
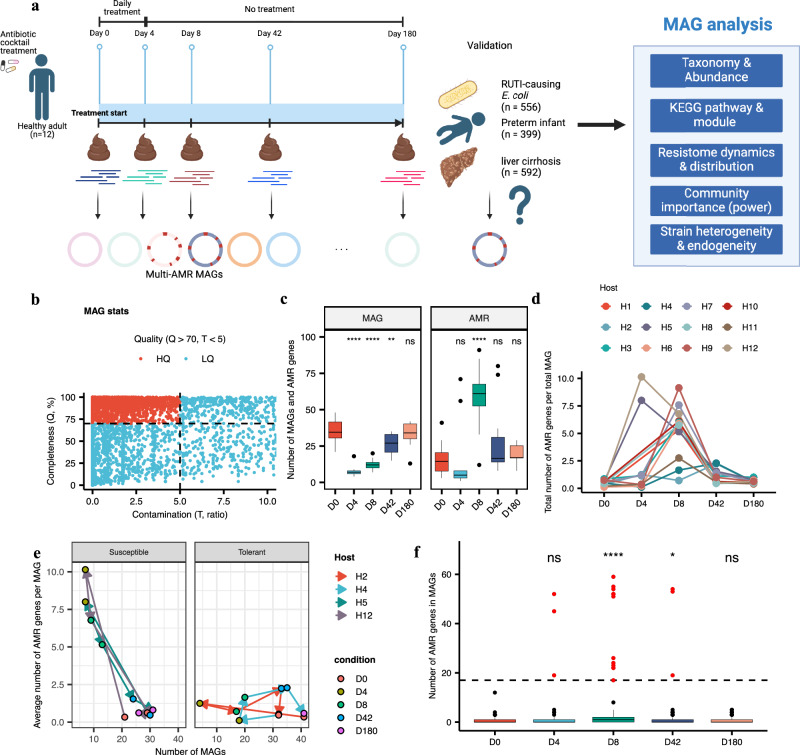
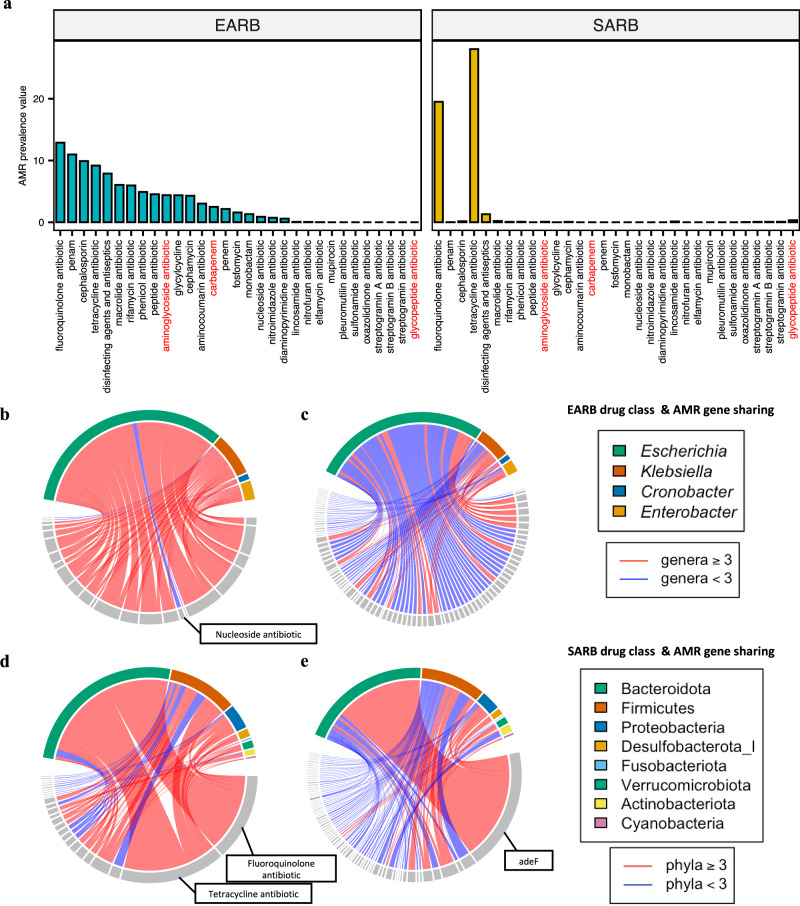
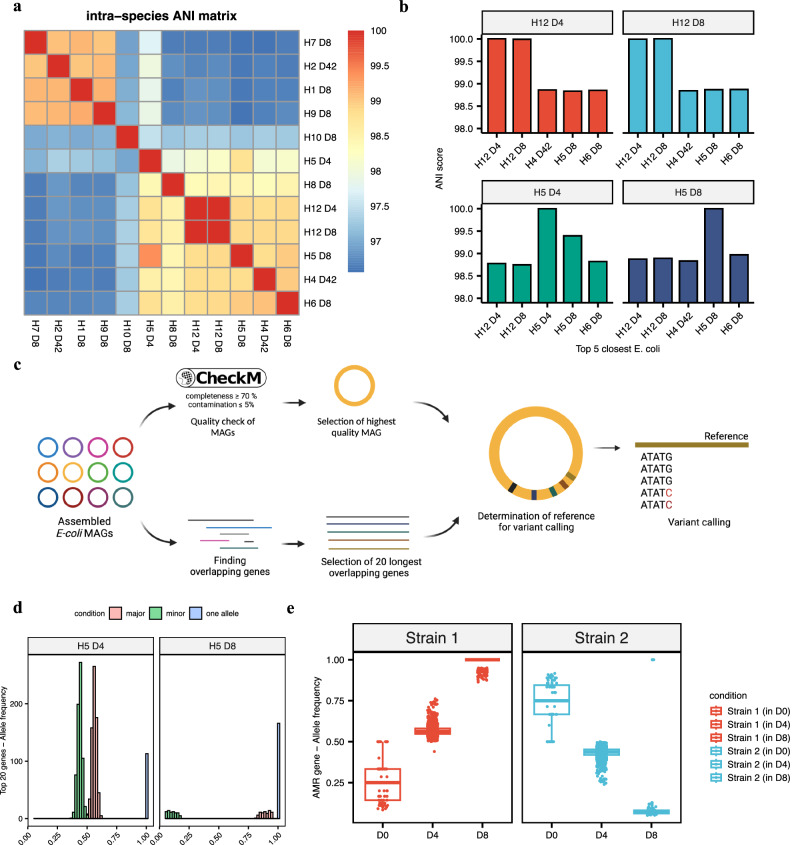
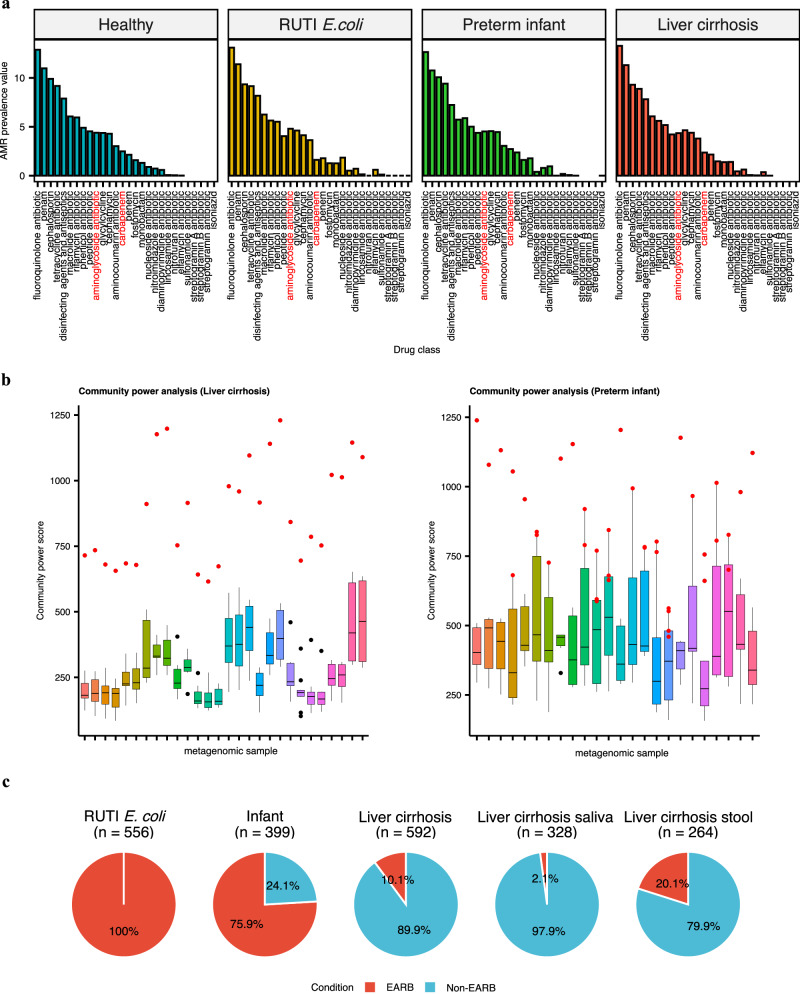
In recent years, the overuse of antibiotics has led to the emergence of antimicrobial-resistant (AMR) bacteria. To evaluate the spread of AMR bacteria, the reservoir of AMR genes (resistome) has been identified in environmental samples, hospital environments, and human populations, but the functional role of AMR bacteria and their persistence within individuals has not been fully investigated. Here, we performed a strain-resolved in-depth analysis of the resistome changes by reconstructing a large number of metagenome-assembled genomes from the gut microbiome of an antibiotic-treated individual. Interestingly, we identified two bacterial populations with different resistome profiles: extensively acquired antimicrobial-resistant bacteria (EARB) and sporadically acquired antimicrobial-resistant bacteria, and found that EARB showed broader drug resistance and a significant functional role in shaping individual microbiome composition after antibiotic treatment. Our findings of AMR bacteria would provide a new avenue for controlling the spread of AMR bacteria in the human community.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: J.W.B., N.P., B.S., N.K., J.N., A.M., S.S., J.-I.K., J.W.S., A.K., and S.L. declared no financial competing interests. A.K. serves as an associate editor of this journal but had no involvement in the peer review or decision-making process for this manuscript. J.N. is a member of the advisory board of the journal, but he had no influence on the peer review process or decision to publish this paper.
Figures


References
MeSH terms
Substances
Grants and funding
- 2021R1C1C1006336, 2021M3A9G8022959, RS-2024-00419699/National Research Foundation of Korea
- 2021R1C1C1006336, 2021M3A9G8022959, RS-2024-00419699/National Research Foundation of Korea
- 2021R1C1C1006336, 2021M3A9G8022959, RS-2024-00419699/National Research Foundation of Korea
- 2021R1C1C1006336, 2021M3A9G8022959, RS-2024-00419699/National Research Foundation of Korea
- 2021R1C1C1006336, 2021M3A9G8022959, RS-2024-00419699/National Research Foundation of Korea
- 2021R1C1C1006336, 2021M3A9G8022959, RS-2024-00419699/National Research Foundation of Korea
- HR22C141105/the Korea Health Industry Development Institute
- HR22C141105/the Korea Health Industry Development Institute
- HR22C141105/the Korea Health Industry Development Institute
- HR22C141105/the Korea Health Industry Development Institute
- HR22C141105/the Korea Health Industry Development Institute
- HR22C141105/the Korea Health Industry Development Institute
- 2024-ER2108-00, 2024-ER0608-00/Korea National Institute of Health
- 2024-ER2108-00, 2024-ER0608-00/Korea National Institute of Health
- 2024-ER2108-00, 2024-ER0608-00/Korea National Institute of Health
- 2024-ER2108-00, 2024-ER0608-00/Korea National Institute of Health
- 2024-ER2108-00, 2024-ER0608-00/Korea National Institute of Health
- 2024-ER2108-00, 2024-ER0608-00/Korea National Institute of Health
- 2024-ER2108-00, 2024-ER0608-00/Korea National Institute of Health
- GIST-MIT research Collaboration grant/GIST Research Institute
- GIST-MIT research Collaboration grant/GIST Research Institute
- GIST-MIT research Collaboration grant/GIST Research Institute
- GIST-MIT research Collaboration grant/GIST Research Institute
- GIST-MIT research Collaboration grant/GIST Research Institute
- GIST-MIT research Collaboration grant/GIST Research Institute
LinkOut - more resources
Full Text Sources
Medical
