

Profiling triple-negative breast cancer-specific super-enhancers identifies high-risk mesenchymal development subtype and BETi-Targetable vulnerabilities
- PMID: 40361105
- PMCID: PMC12070678
- DOI: 10.1186/s12943-025-02342-6
Profiling triple-negative breast cancer-specific super-enhancers identifies high-risk mesenchymal development subtype and BETi-Targetable vulnerabilities
Abstract
Background: Super-enhancers (SEs) are critical regulators of tumorigenesis and represent promising targets for bromodomain and extra-terminal domain inhibitors (BETi). However, clinical studies across various solid tumors, including triple-negative breast cancer (TNBC), have demonstrated limited BETi efficacy. This study aims to investigate SE heterogeneity in TNBC and its influence on BETi effectiveness, with the goal of advancing BETi precision treatment strategies and enhancing therapeutic efficacy.
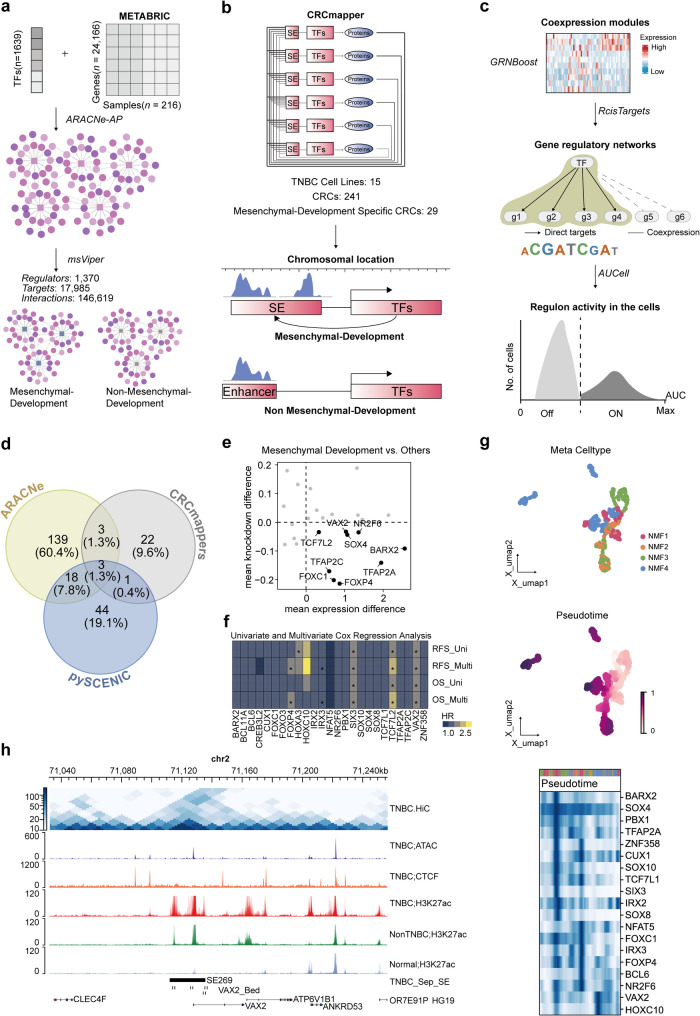
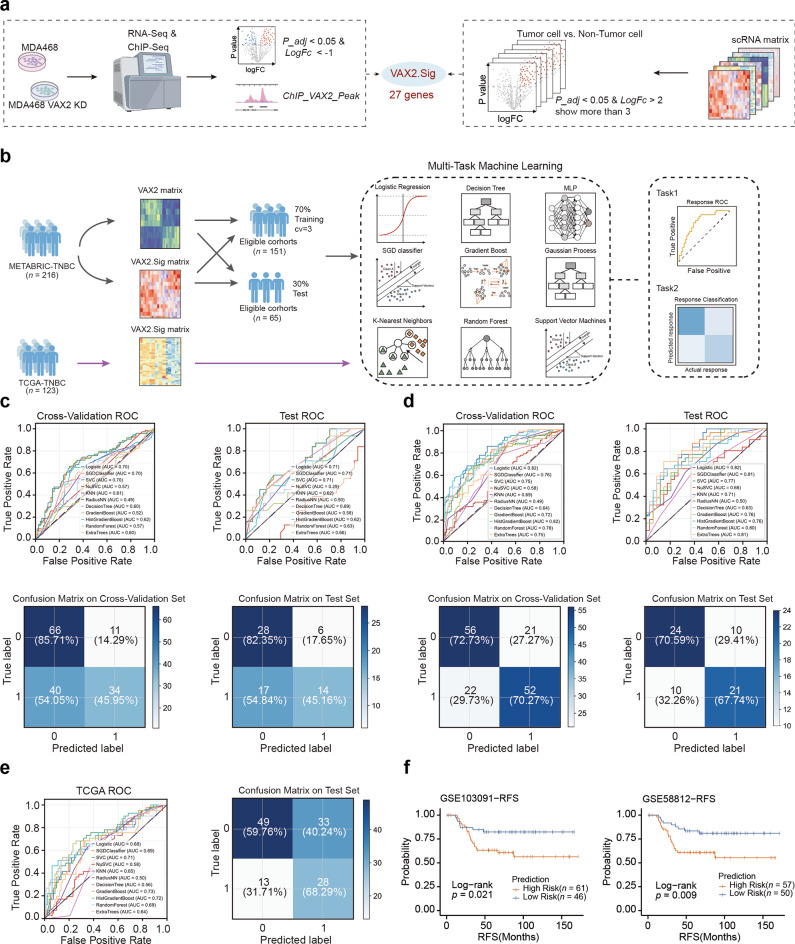
Methods: We conducted a comprehensive analysis of H3K27ac ChIP-Seq data from TNBC cell lines and clinical samples, integrating multiple bulk RNA-Seq, scRNA-Seq, and stRNA-Seq datasets to characterize the SE landscape and heterogeneity in TNBC. Utilizing various bioinformatics algorithms, CERES scoring, and clinical prognostic data on transcription factors (TFs), we identified core transcriptional regulatory circuits (CRCs) composed of TNBC-specific SEs and master regulators, characterizing different TNBC subtypes. The biological significance of CRCs in these different TNBC subtypes and their influence on BETi sensitivity were evaluated using in vitro and in vivo models.
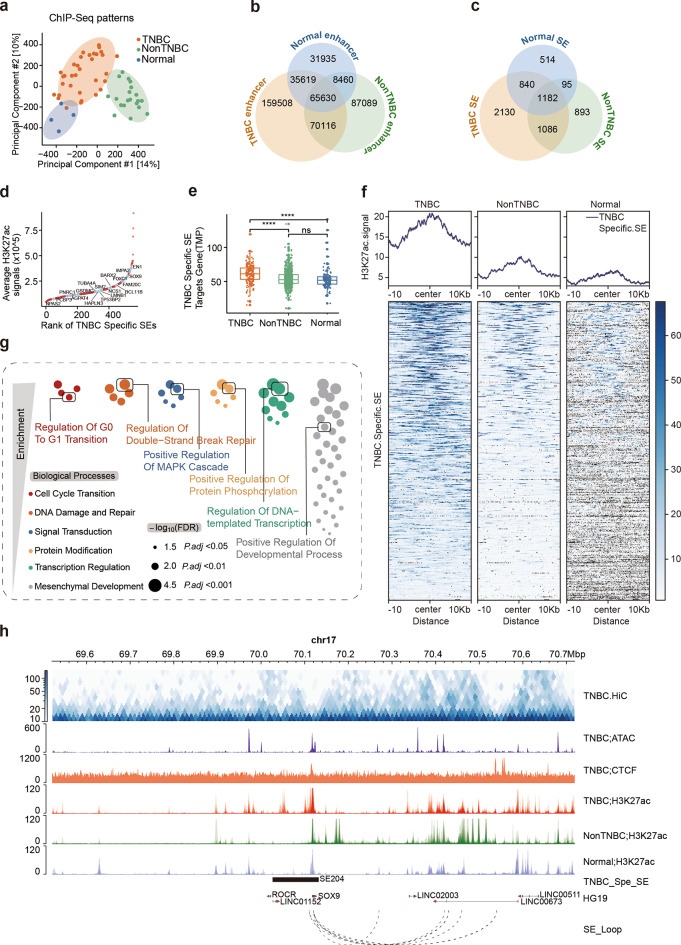
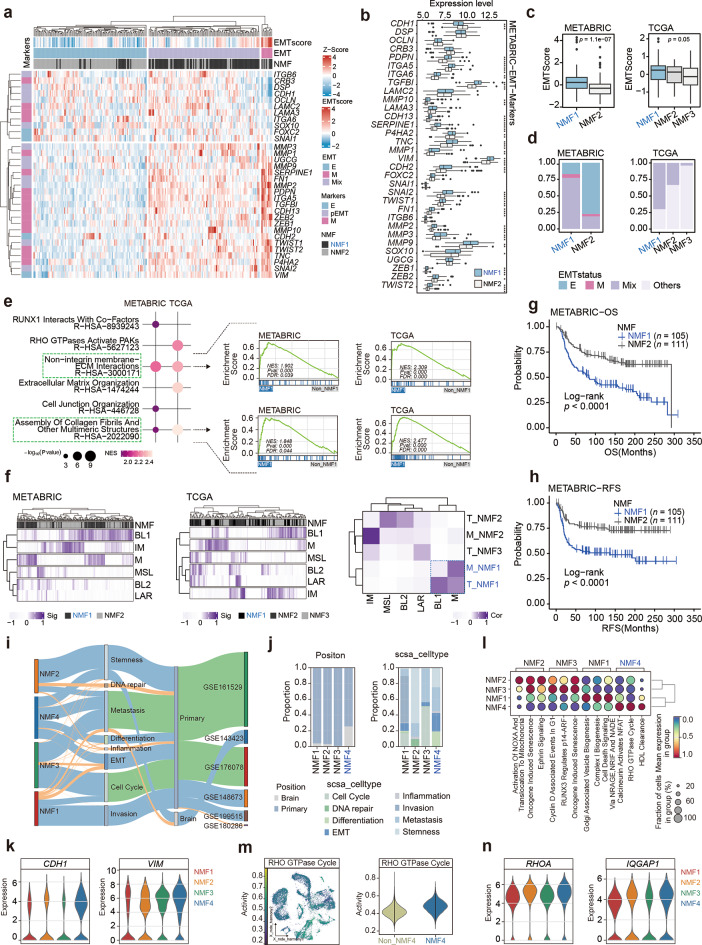
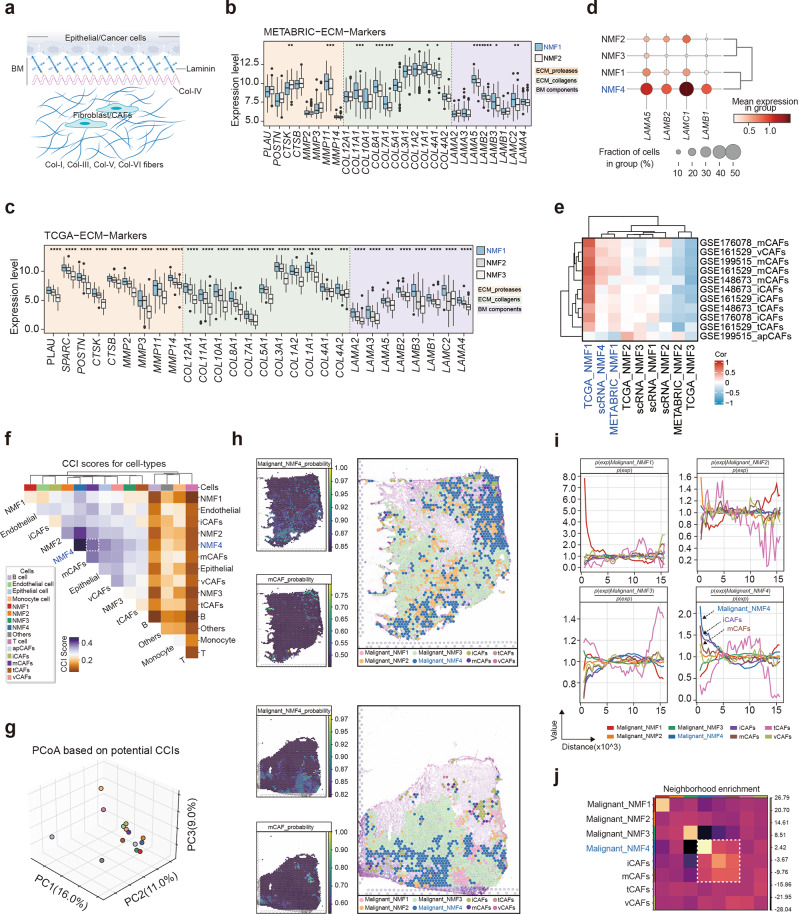
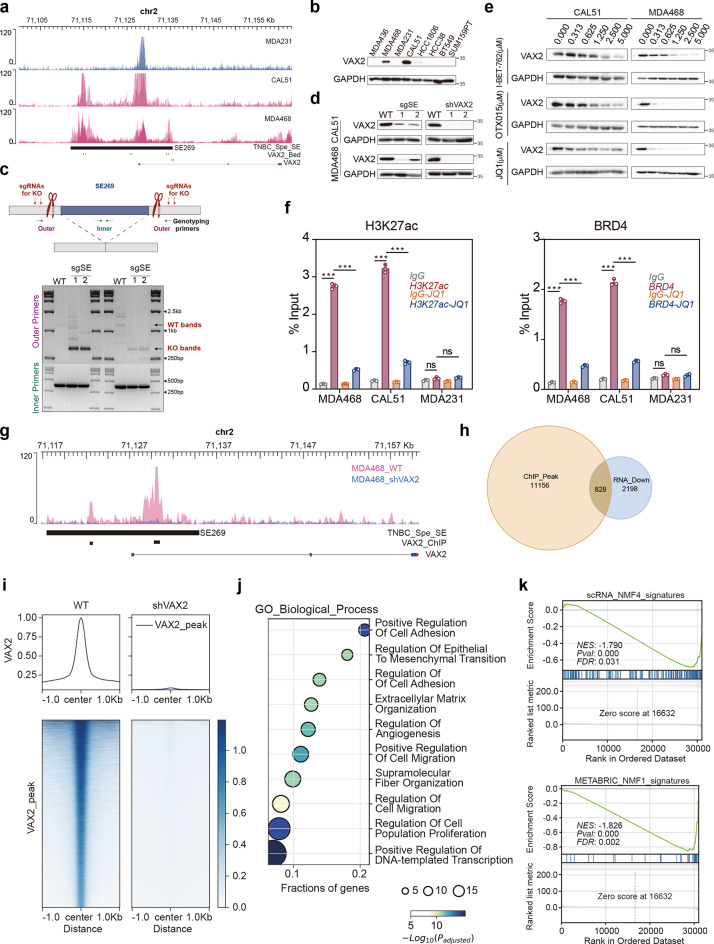
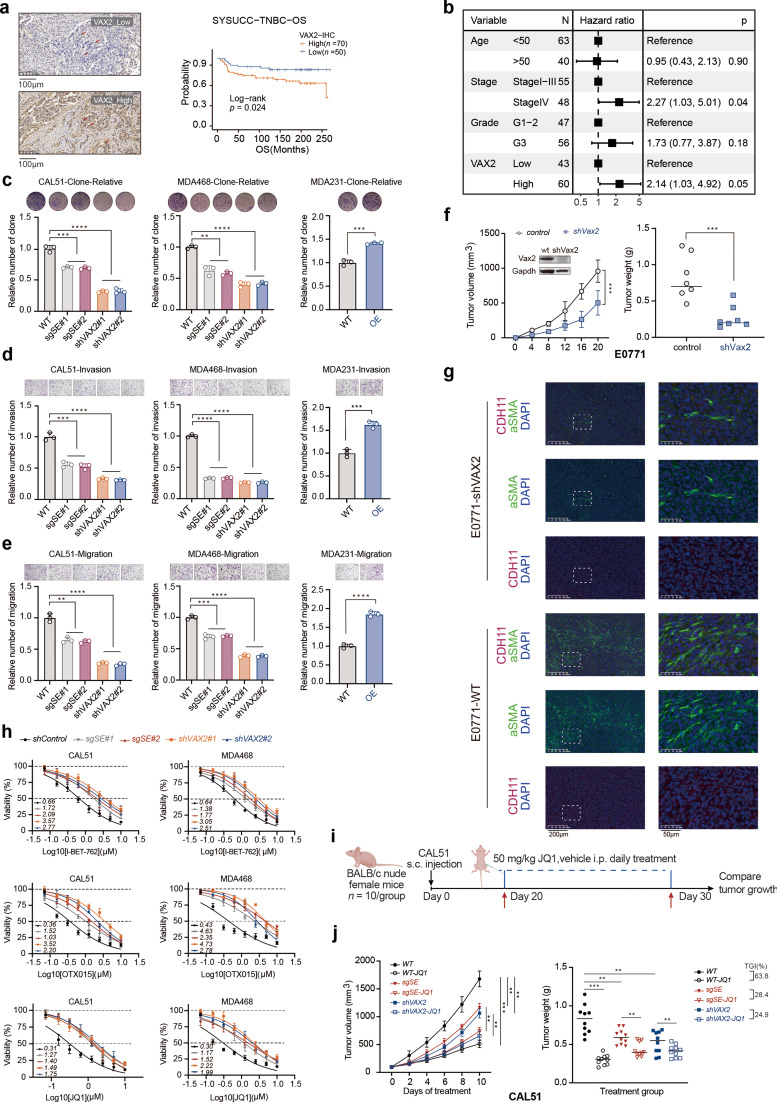
Results: Our findings revealed a distinct SE landscape in TNBC compared to non-TNBC and normal breast epithelium, allowing TNBC to be classified into distinct subtypes based on TNBC-specific SEs. Importantly, we identified a high-risk mesenchymal development subtype, validated across cell lines and transcriptomic analyses, primarily driven by a CRC consisting of the master regulator VAX2 and a TNBC-specific SE. This SE-VAX2 CRC is essential for sustaining the malignant traits of this subtype and increasing its sensitivity to BETi.
Conclusions: Our research clarifies the heterogeneity of SEs in TNBC and identifies a high-risk mesenchymal development subtype driven by the SE-VAX2 CRC. The subtype shows more sensitivity to BETi, supporting its precision application in TNBC.
Keywords: Heterogeneity; Super-enhancers; Transcription factors; Triple-negative breast cancer.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethical approval: The study protocol was approved by the Medical Ethics Committee of Sun Yat-sen University Cancer Center (Medical Research Ethics Review No. G2021-092-01). The study conformed to the principles of the Helsinki Declaration. Competing interests: The authors declare no competing interests.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
