

Alzheimer's Is a Multiform Disease of Sustained Neuronal Integrated Stress Response Driven by the C99 Fragment Generated Independently of AβPP; Proteolytic Production of Aβ Is Suppressed in AD-Affected Neurons: Evolution of a Theory
- PMID: 40362488
- PMCID: PMC12073115
- DOI: 10.3390/ijms26094252
Alzheimer's Is a Multiform Disease of Sustained Neuronal Integrated Stress Response Driven by the C99 Fragment Generated Independently of AβPP; Proteolytic Production of Aβ Is Suppressed in AD-Affected Neurons: Evolution of a Theory
Abstract
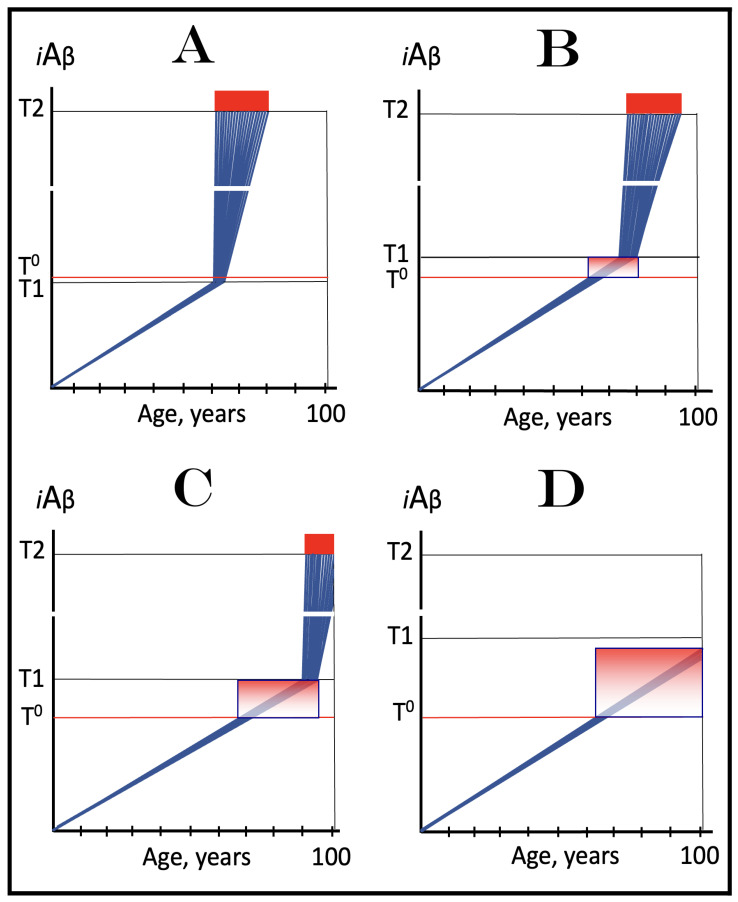
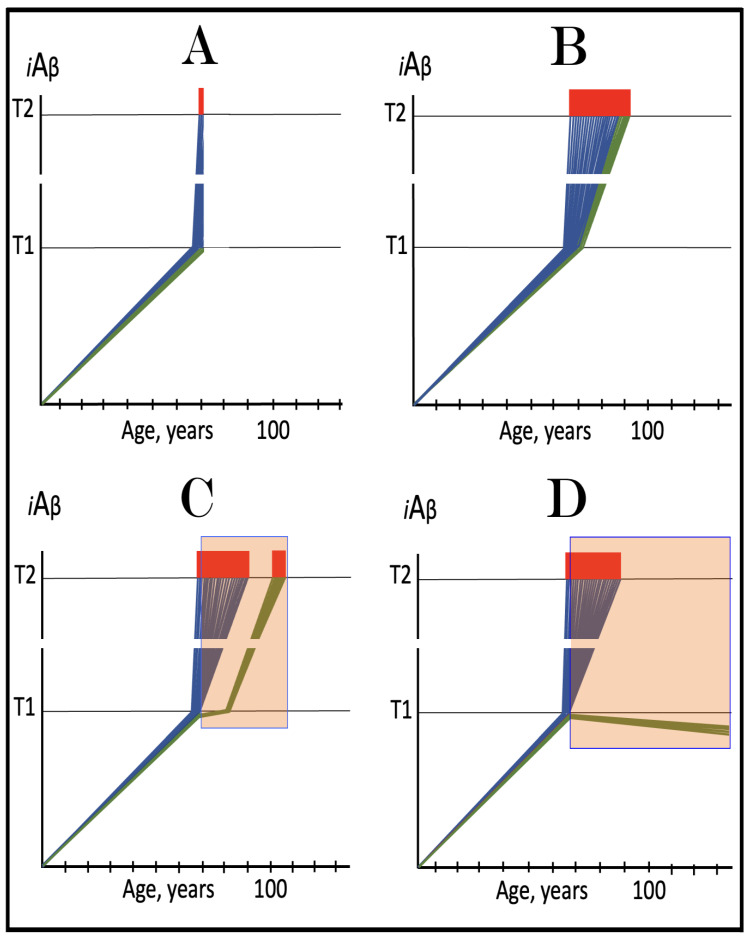
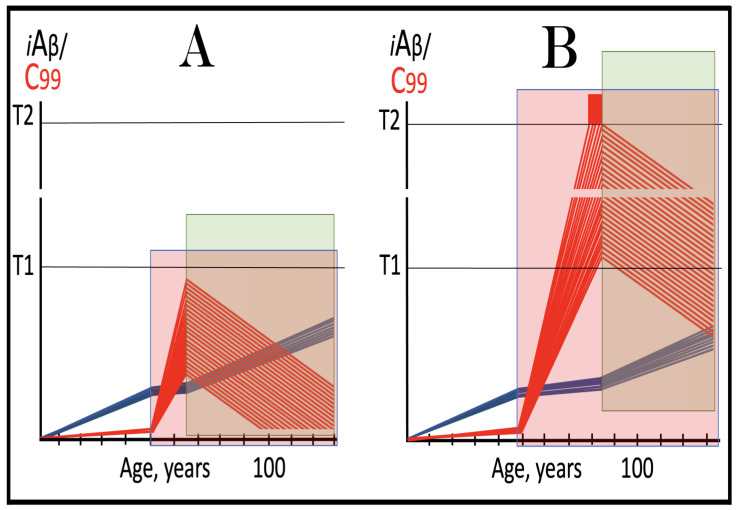
The present Perspective analyzes the remarkable evolution of the Amyloid Cascade Hypothesis 2.0 (ACH2.0) theory of Alzheimer's disease (AD) since its inception a few years ago, as reflected in the diminishing role of amyloid-beta (Aβ) in the disease. In the initial iteration of the ACH2.0, Aβ-protein-precursor (AβPP)-derived intraneuronal Aβ (iAβ), accumulated to neuronal integrated stress response (ISR)-eliciting levels, triggers AD. The neuronal ISR, in turn, activates the AβPP-independent production of its C99 fragment that is processed into iAβ, which drives the disease. The second iteration of the ACH2.0 stemmed from the realization that AD is, in fact, a disease of the sustained neuronal ISR. It introduced two categories of AD-conventional and unconventional-differing mainly in the manner of their causation. The former is caused by the neuronal ISR triggered by AβPP-derived iAβ, whereas in the latter, the neuronal ISR is elicited by stressors distinct from AβPP-derived iAβ and arising from brain trauma, viral and bacterial infections, and various types of inflammation. Moreover, conventional AD always contains an unconventional component, and in both forms, the disease is driven by iAβ generated independently of AβPP. In its third, the current, iteration, the ACH2.0 posits that proteolytic production of Aβ is suppressed in AD-affected neurons and that the disease is driven by C99 generated independently of AβPP. Suppression of Aβ production in AD seems an oxymoron: Aβ is equated with AD, and the later is inconceivable without the former in an ingrained Amyloid Cascade Hypothesis (ACH)-based notion. But suppression of Aβ production in AD-affected neurons is where the logic leads, and to follow it we only need to overcome the inertia of the preexisting assumptions. Moreover, not only is the generation of Aβ suppressed, so is the production of all components of the AβPP proteolytic pathway. This assertion is not a quantum leap (unless overcoming the inertia counts as such): the global cellular protein synthesis is severely suppressed under the neuronal ISR conditions, and there is no reason for constituents of the AβPP proteolytic pathway to be exempted, and they, apparently, are not, as indicated by the empirical data. In contrast, tau protein translation persists in AD-affected neurons under ISR conditions because the human tau mRNA contains an internal ribosomal entry site in its 5'UTR. In current mouse models, iAβ derived from AβPP expressed exogenously from human transgenes elicits the neuronal ISR and thus suppresses its own production. Its levels cannot principally reach AD pathology-causing levels regardless of the number of transgenes or the types of FAD mutations that they (or additional transgenes) carry. Since the AβPP-independent C99 production pathway is inoperative in mice, the current transgenic models have no potential for developing the full spectrum of AD pathology. What they display are only effects of the AβPP-derived iAβ-elicited neuronal ISR. The paper describes strategies to construct adequate transgenic AD models. It also details the utilization of human neuronal cells as the only adequate model system currently available for conventional and unconventional AD. The final alteration of the ACH2.0, introduced in the present Perspective, is that AβPP, which supports neuronal functionality and viability, is, after all, potentially produced in AD-affected neurons, albeit not conventionally but in an ISR-driven and -compatible process. Thus, the present narrative begins with the "omnipotent" Aβ capable of both triggering and driving the disease and ends up with this peptide largely dislodged from its pedestal and retaining its central role in triggering the disease in only one, although prevalent (conventional), category of AD (and driving it in none). Among interesting inferences of the present Perspective is the determination that "sporadic AD" is not sporadic at all ("non-familial" would be a much better designation). The term has fatalistic connotations, implying that the disease can strike at random. This is patently not the case: The conventional disease affects a distinct subpopulation, and the basis for unconventional AD is well understood. Another conclusion is that, unless prevented, the occurrence of conventional AD is inevitable given a sufficiently long lifespan. This Perspective also defines therapeutic directions not to be taken as well as auspicious ways forward. The former category includes ACH-based drugs (those interfering with the proteolytic production of Aβ and/or depleting extracellular Aβ). They are legitimate (albeit inefficient) preventive agents for conventional AD. There is, however, a proverbial snowball's chance in hell of them being effective in symptomatic AD, lecanemab, donanemab, and any other "…mab" or "…stat" notwithstanding. They comprise Aβ-specific antibodies, inhibitors of beta- and gamma-secretase, and modulators of the latter. In the latter category, among ways to go are the following: (1) Depletion of iAβ, which, if sufficiently "deep", opens up a tantalizing possibility of once-in-a-lifetime preventive transient treatment for conventional AD and aging-associated cognitive decline, AACD. (2) Composite therapy comprising the degradation of C99/iAβ and concurrent inhibition of the neuronal ISR. A single transient treatment could be sufficient to arrest the progression of conventional AD and prevent its recurrence for life. Multiple recurrent treatments would achieve the same outcome in unconventional AD. Alternatively, the sustained reduction/removal of unconventional neuronal ISR-eliciting stressors through the elimination of their source would convert unconventional AD into conventional one, preventable/treatable by a single transient administration of the composite C99/iAβ depletion/ISR suppression therapy. Efficient and suitable ISR inhibitors are available, and it is explicitly clear where to look for C99/iAβ-specific targeted degradation agents-activators of BACE1 and, especially, BACE2. Directly acting C99/iAβ-specific degradation agents such as proteolysis-targeting chimeras (PROTACs) and molecular-glue degraders (MGDs) are also viable options. (3) A circumscribed shift (either upstream or downstream) of the position of transcription start site (TSS) of the human AβPP gene, or, alternatively, a gene editing-mediated excision or replacement of a small, defined segment of its portion encoding 5'-untranslated region of AβPP mRNA; targeting AβPP RNA with anti-antisense oligonucleotides is another possibility. If properly executed, these RNA-based strategies would not interfere with the protein-coding potential of AβPP mRNA, and each would be capable of both preventing and stopping the AβPP-independent generation of C99 and thus of either preventing AD or arresting the progression of the disease in its conventional and unconventional forms. The paper is interspersed with "validation" sections: every conceptually significant notion is either validated by the existing data or an experimental procedure validating it is proposed.
Keywords: AD as the disease of the neuronal ISR; Amyloid Cascade Hypothesis 2.0 (ACH2.0); AβPP-independent generation of the C99 fragment; C99 as the driver of AD; ISR-mediated suppression of the AβPP proteolytic pathway in AD-affected neurons; RNA-based AD therapies; RNA-dependent amplification of human AβPP mRNA; concurrent inhibition of the neuronal ISR and activation of BACE1 and BACE2 as composite AD therapy; conventional and unconventional Alzheimer’s disease (AD); inhibition of the neuronal ISR as AD therapy; neuronal integrated stress response (ISR).
Conflict of interest statement
The authors declare no conflicts of interest.
Figures












































References
-
- Volloch V., Rits-Volloch S. Principles of Design of Clinical Trials for Prevention and Treatment of Alzheimer’s Disease and Aging-Associated Cognitive Decline in the ACH2.0 Perspective: Potential Outcomes, Challenges and Solutions. J. Alzheimers Dis. Rep. 2023;7:921–955. doi: 10.3233/ADR-230037. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
