

Lipopolysaccharide-induced DNA damage response activates DNA-PKcs to drive actin cytoskeleton disruption and cardiac microvascular dysfunction in endotoxemia
- PMID: 40365284
- PMCID: PMC12068286
- DOI: 10.7150/thno.111266
Lipopolysaccharide-induced DNA damage response activates DNA-PKcs to drive actin cytoskeleton disruption and cardiac microvascular dysfunction in endotoxemia
Abstract
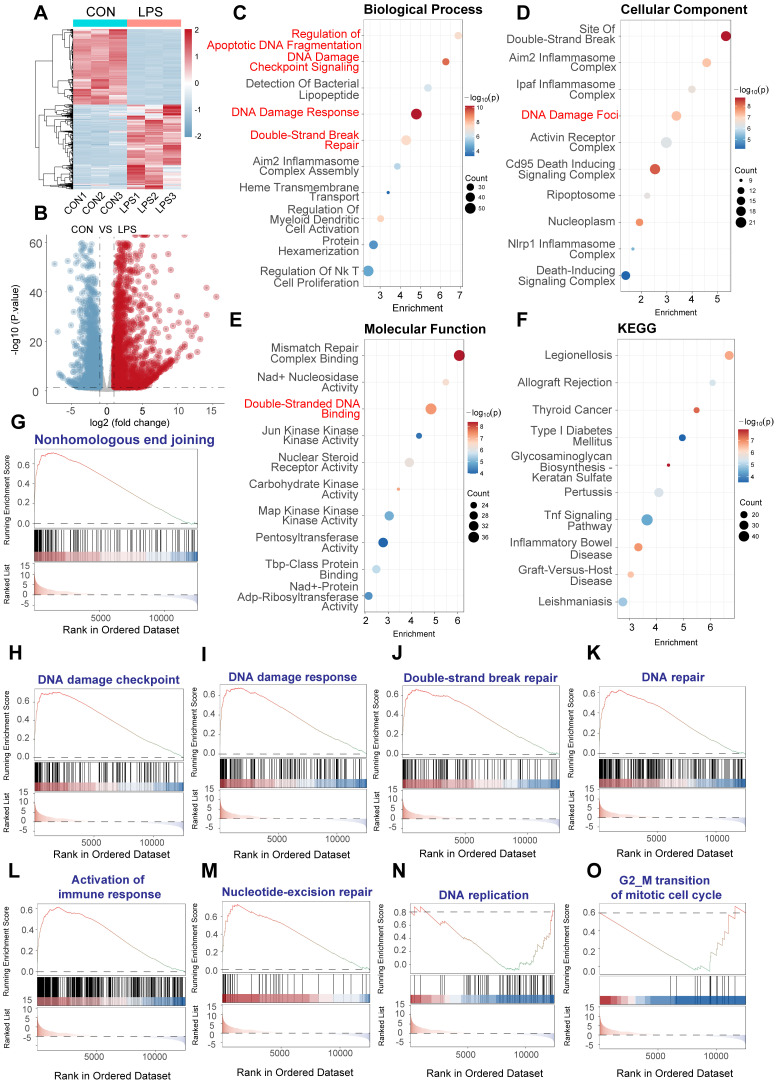
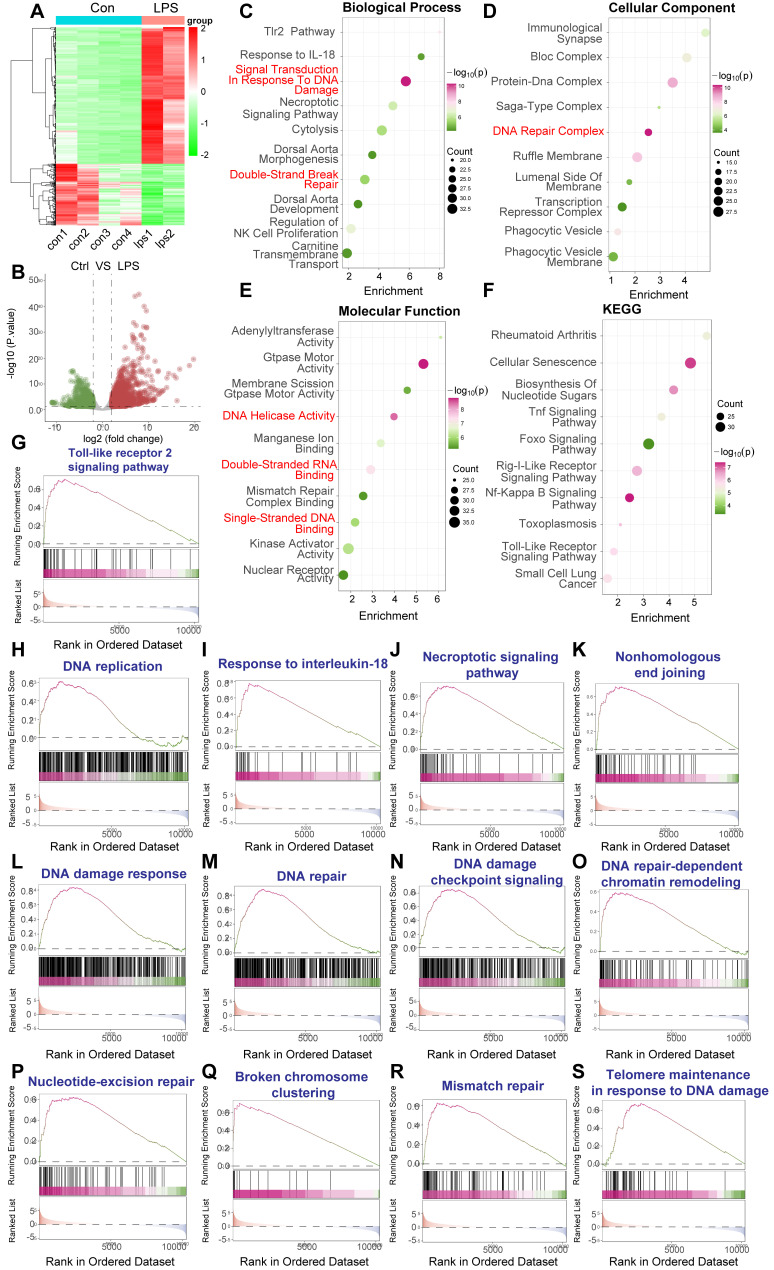
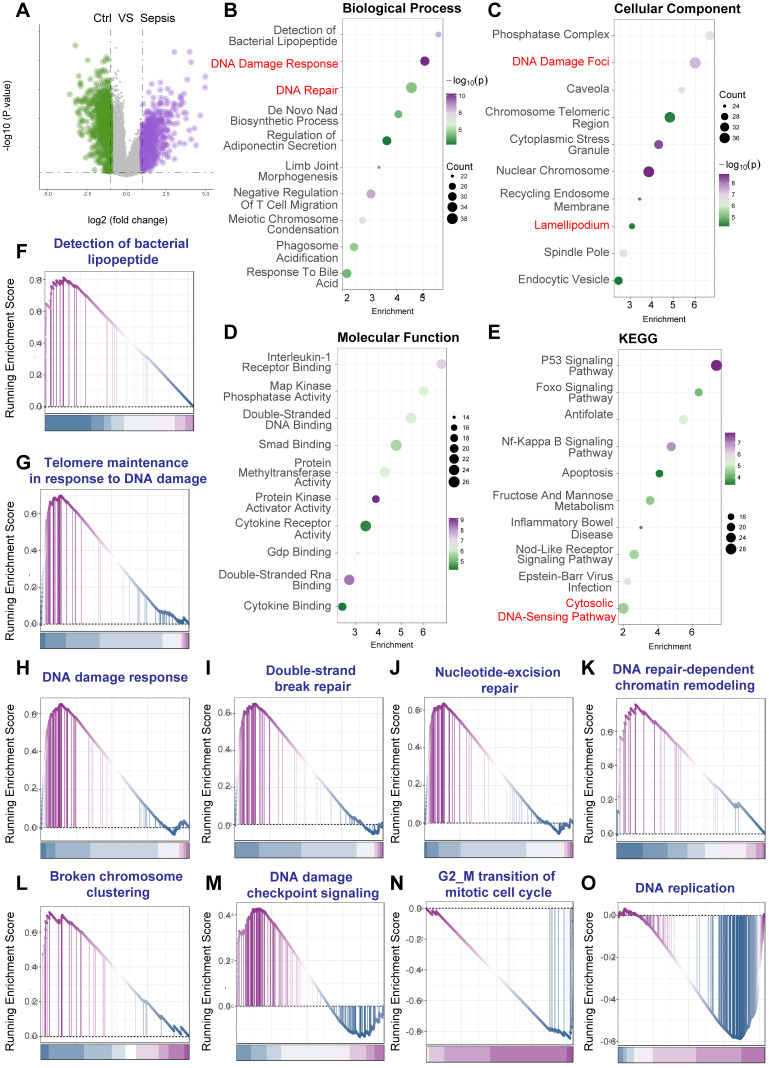
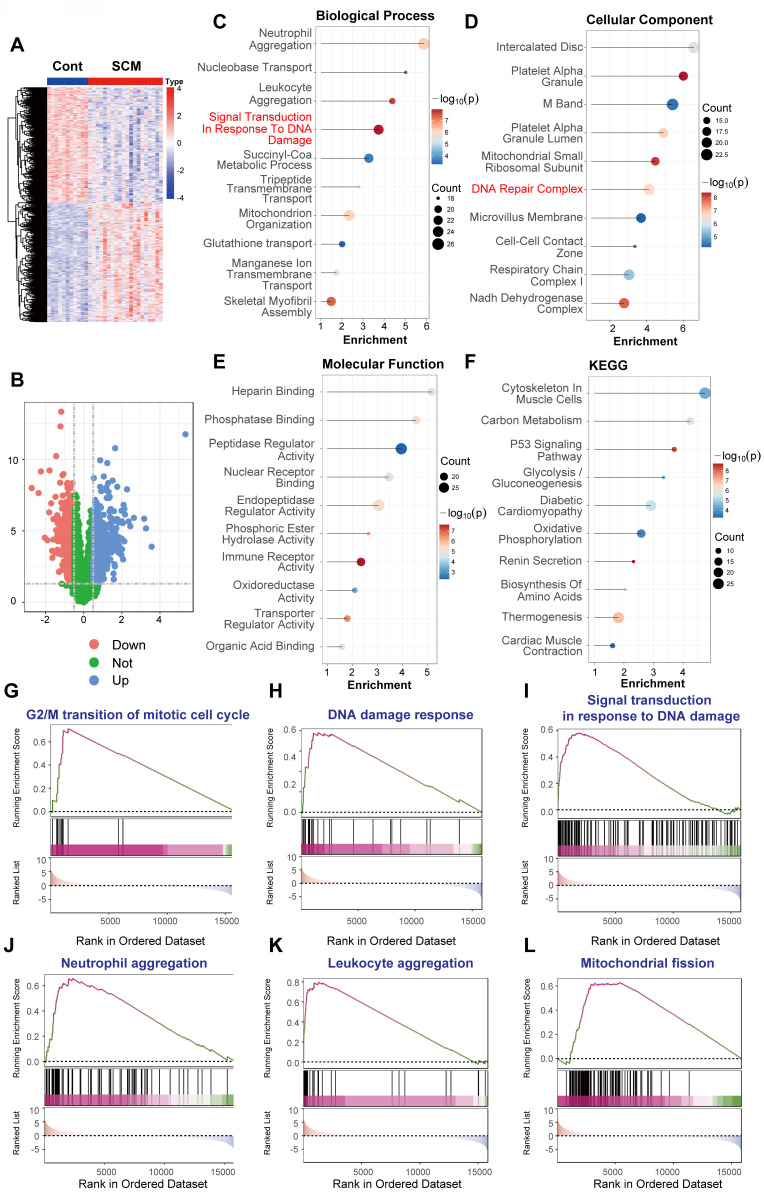
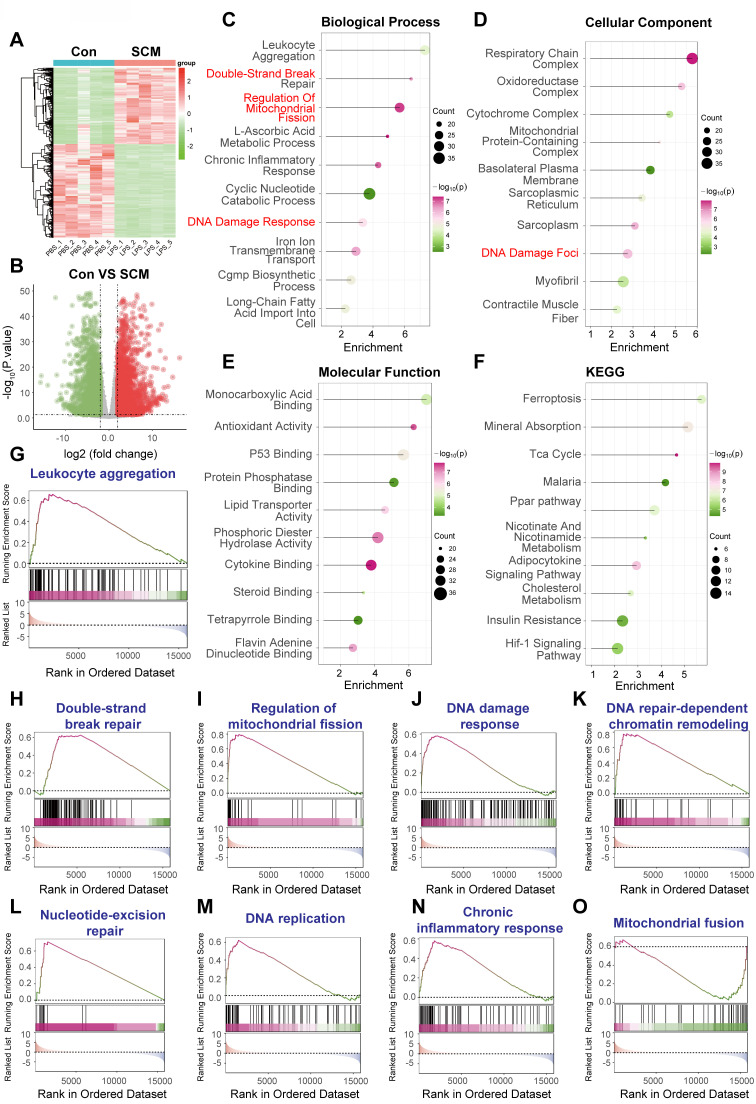
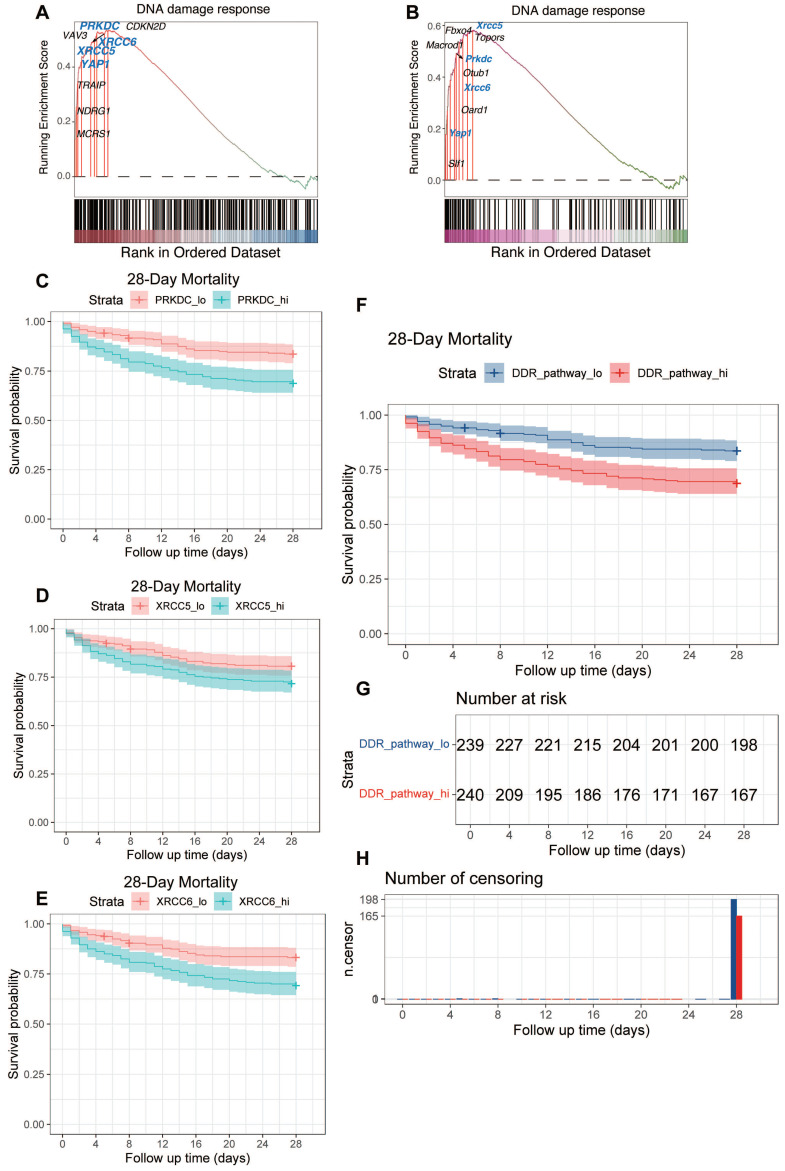
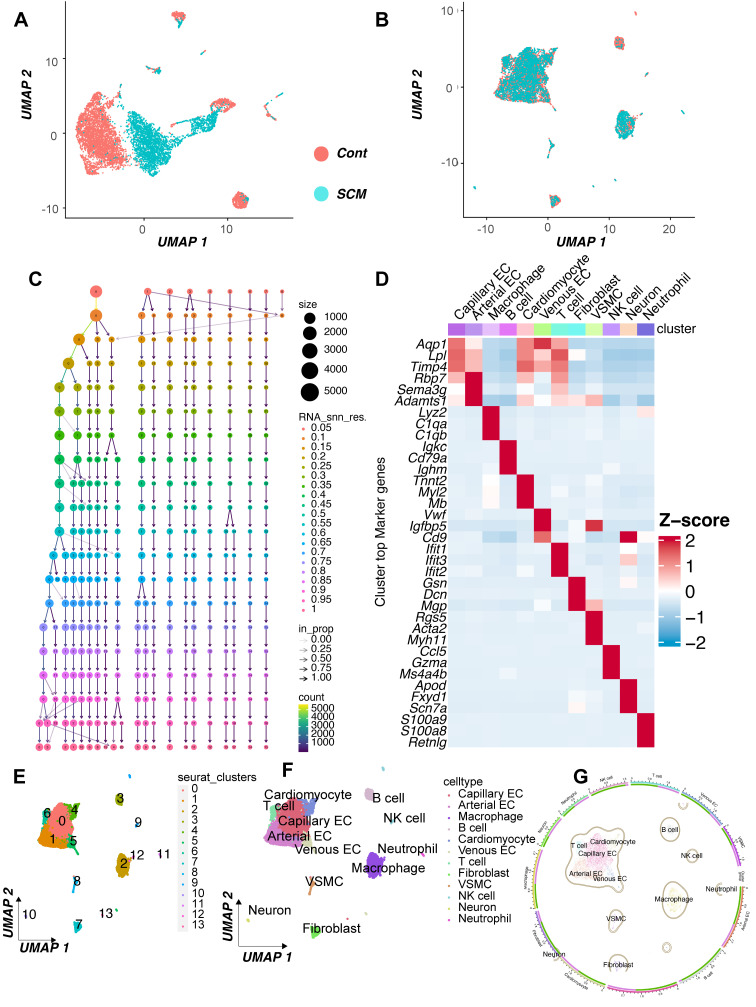
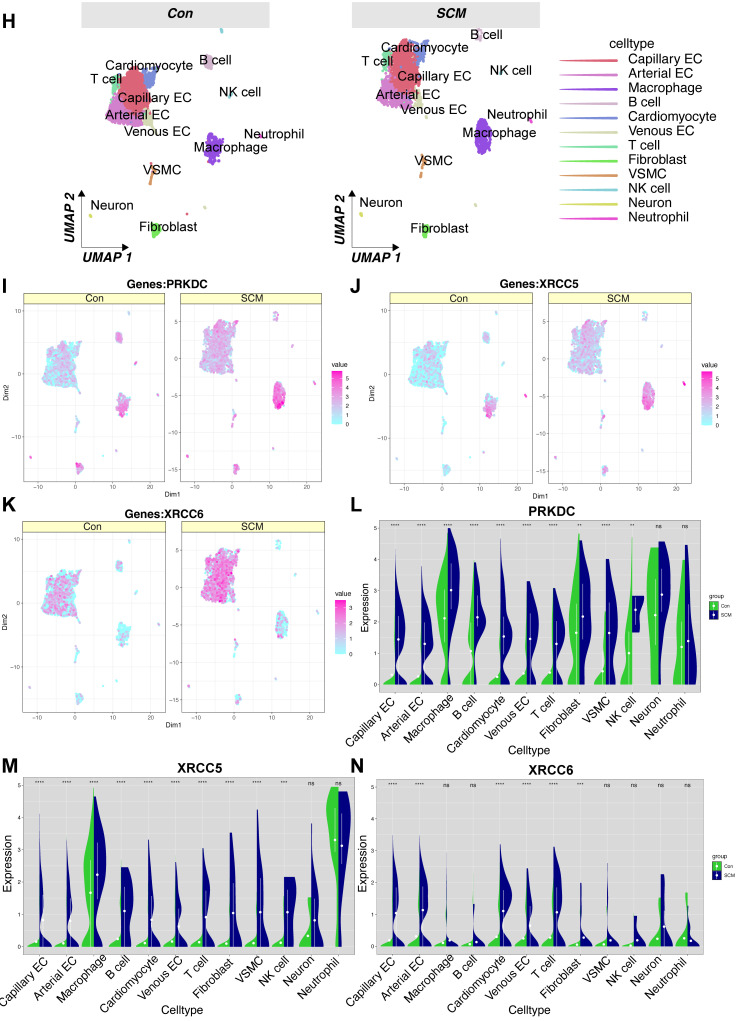
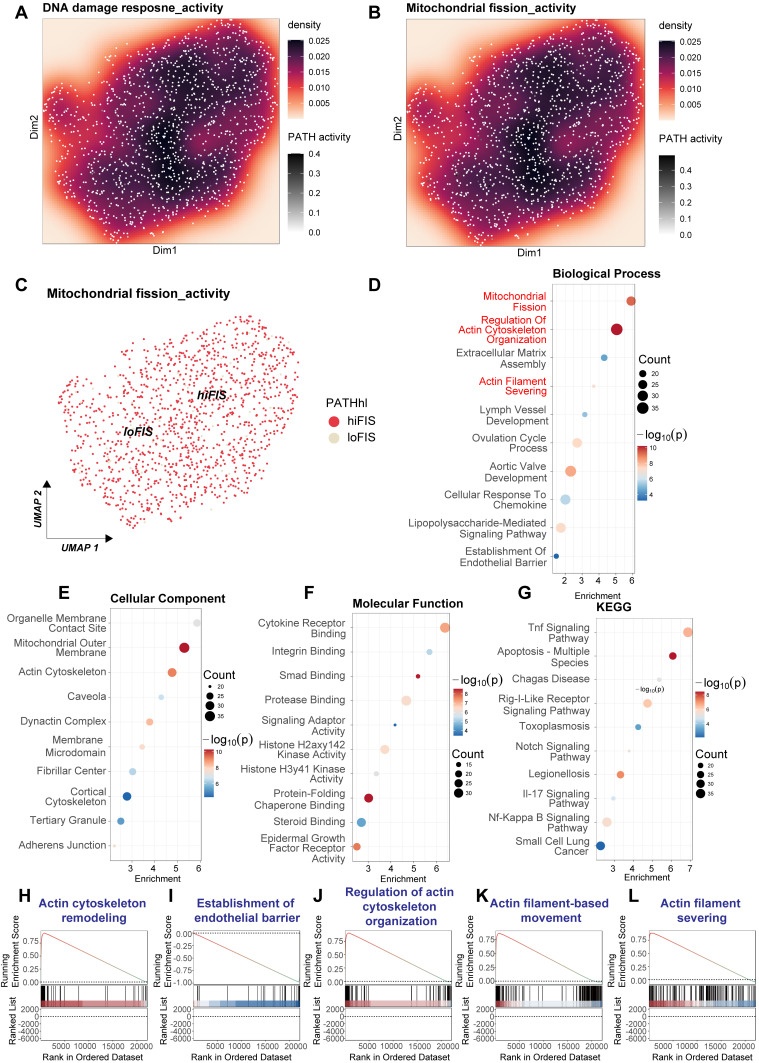
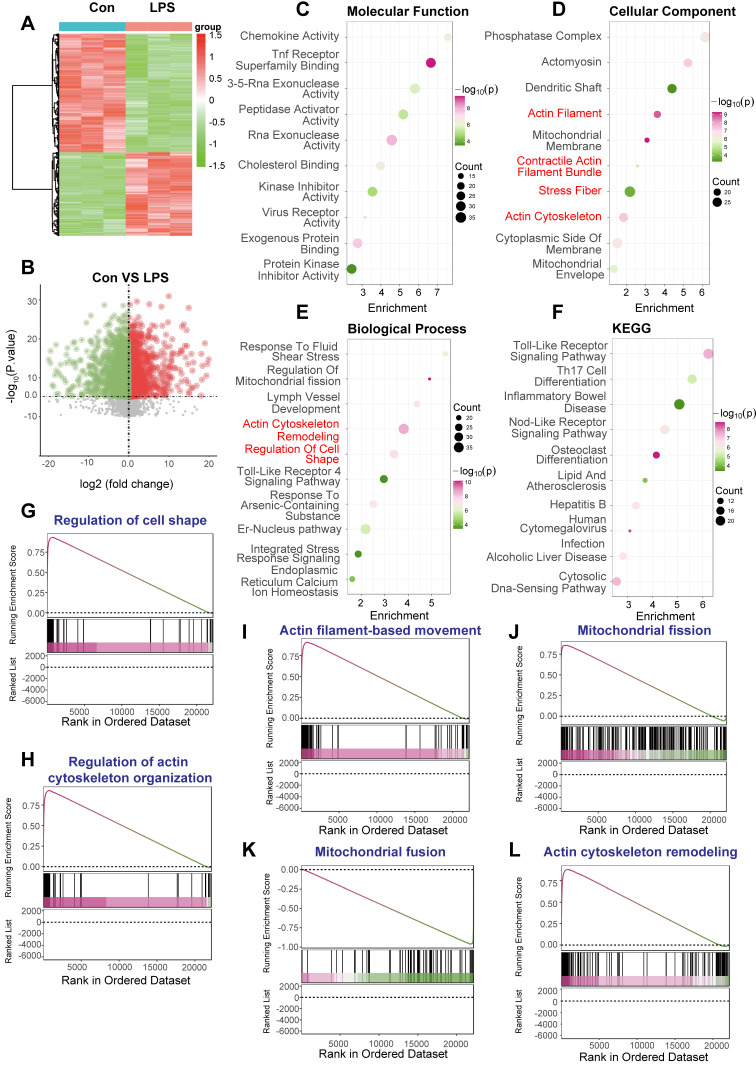
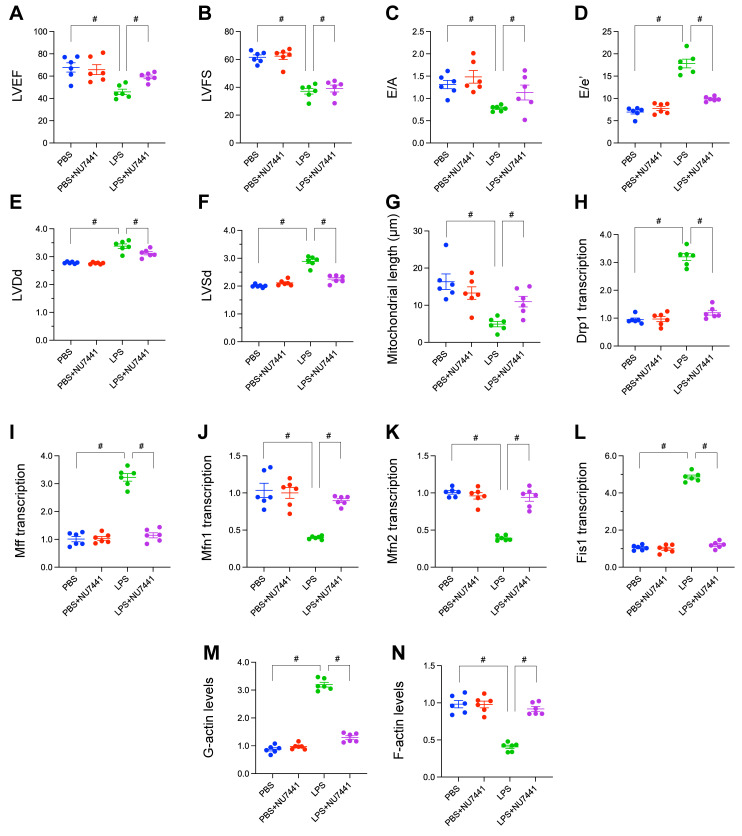
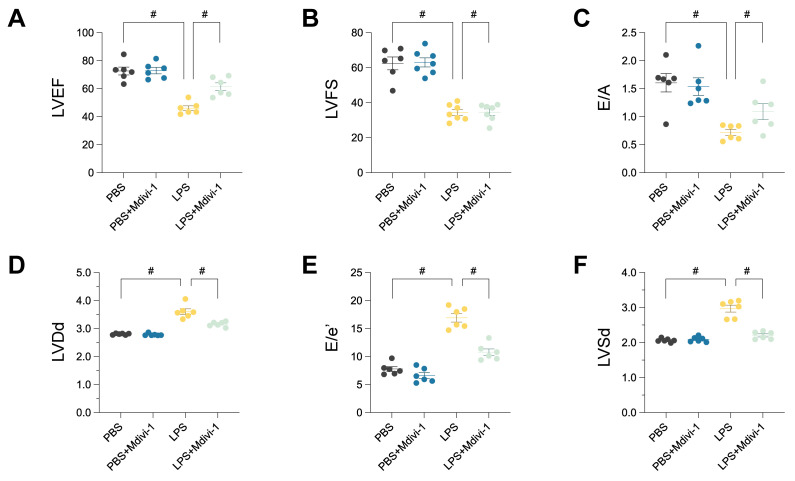
Rationale: Sepsis-induced cardiomyopathy is characterized by microvascular injury, which is linked to lipopolysaccharide (LPS)-induced DNA damage response (DDR). This study investigates the role of DNA-PKcs, a key enzyme in the DDR pathway, in driving actin disruption and microvascular dysfunction following LPS exposure. Methods: We analyzed diverse transcriptomic datasets from septic human and murine models using bioinformatics tools to assess DDR pathway activation, correlations, and prognosis. In vivo, LPS-challenged mice were treated with inhibitors of DNA-PKcs or mitochondrial fission, and we evaluated cardiac function, microvascular integrity, mitochondrial status, and actin polymerization. Results: Bioinformatic analyses consistently revealed significant activation of the DDR pathway and upregulation of key genes across diverse septic models. Notably, elevated DDR pathway activity was significantly correlated with poor 28-day survival in human sepsis patients. Single-cell analysis localized this DDR gene upregulation predominantly to cardiac endothelial cells (ECs), fibroblasts, and macrophages during sepsis. Within septic capillary ECs, DDR pathway activity scores strongly correlated spatially and functionally with heightened mitochondrial fission and cytoskeletal remodeling pathway activities. In vivo experiments confirmed that LPS induced severe systolic and diastolic dysfunction, microvascular damage, and mitochondrial fragmentation, as well as significant actin depolymerization. Inhibition of DNA-PKcs with NU7441 markedly attenuated all these LPS-induced pathologies, improving cardiac function, preserving microvascular structure, preventing mitochondrial fragmentation, and normalizing related gene expression and actin cytoskeleton stability. Additionally, inhibiting mitochondrial fission with Mdivi-1 significantly ameliorated LPS-induced cardiac dysfunction and microvascular injury. Conclusions: Our findings suggest that LPS triggers a DNA-PKcs-dependent DDR that promotes mitochondrial fragmentation and actin disruption, particularly in cardiac ECs, contributing to sepsis-induced cardiomyopathy. Targeting DNA-PKcs or mitochondrial fission may hold therapeutic potential for the treatment of sepsis-induced cardiomyopathy.
Keywords: DNA damage response; DNA-PKcs; actin; cytoskeleton; mitochondria.
© The author(s).
Conflict of interest statement
Competing interests: The authors have declared that no competing interest exists.
Figures
Similar articles
-
The DNA-dependent protein kinase catalytic subunit exacerbates endotoxemia-induced myocardial microvascular injury by disrupting the MOTS-c/JNK pathway and inducing profilin-mediated lamellipodia degradation.Theranostics. 2024 Feb 4;14(4):1561-1582. doi: 10.7150/thno.92650. eCollection 2024. Theranostics. 2024. PMID: 38389837 Free PMC article.
-
The DNA-dependent protein kinase catalytic subunit promotes sepsis-induced cardiac dysfunction through disrupting INF-2-dependent mitochondrial dynamics.Int J Med Sci. 2024 Feb 12;21(4):714-724. doi: 10.7150/ijms.91894. eCollection 2024. Int J Med Sci. 2024. PMID: 38464839 Free PMC article.
-
Endotoxemia Engages the RhoA Kinase Pathway to Impair Cardiac Function By Altering Cytoskeleton, Mitochondrial Fission, and Autophagy.Antioxid Redox Signal. 2016 Apr 1;24(10):529-42. doi: 10.1089/ars.2015.6421. Epub 2016 Jan 7. Antioxid Redox Signal. 2016. PMID: 26602979
-
DNA-PKcs Phosphorylates Cofilin2 to Induce Endothelial Dysfunction and Microcirculatory Disorder in Endotoxemic Cardiomyopathy.Research (Wash D C). 2024 Mar 26;7:0331. doi: 10.34133/research.0331. eCollection 2024. Research (Wash D C). 2024. PMID: 38550779 Free PMC article.
-
Nuclear damage-induced DNA damage response coupled with IFI16-driven ECM remodeling underlies dilated cardiomyopathy.Theranostics. 2025 Apr 28;15(12):5998-6021. doi: 10.7150/thno.112247. eCollection 2025. Theranostics. 2025. PMID: 40365289 Free PMC article.
References
-
- Qiao W, Huang Y, Bian Z, Sun X, Wang X, Gao Q. et al. Lipopolysaccharide-induced DNA damage response activates nuclear factor κB signalling pathway via GATA4 in dental pulp cells. Int Endod J. 2019;52:1704–15. - PubMed
-
- Burma S, Chen DJ. Role of DNA-PK in the cellular response to DNA double-strand breaks. DNA Repair (Amst) 2004;3:909–18. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
