

GPU Accelerated Hybrid Particle-Field Molecular Dynamics: Multi-Node/Multi-GPU Implementation and Large-Scale Benchmarks of the OCCAM Code
- PMID: 40365831
- PMCID: PMC12076535
- DOI: 10.1002/jcc.70126
GPU Accelerated Hybrid Particle-Field Molecular Dynamics: Multi-Node/Multi-GPU Implementation and Large-Scale Benchmarks of the OCCAM Code
Abstract
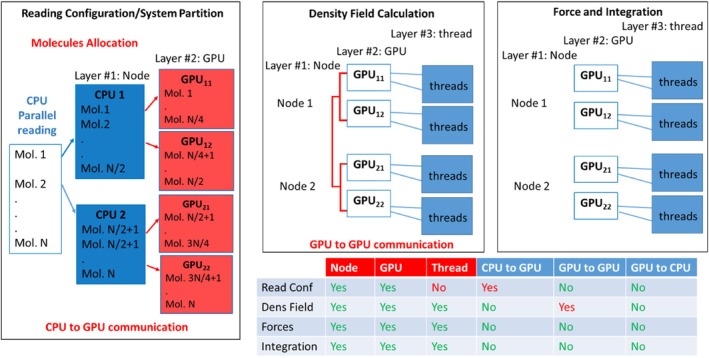
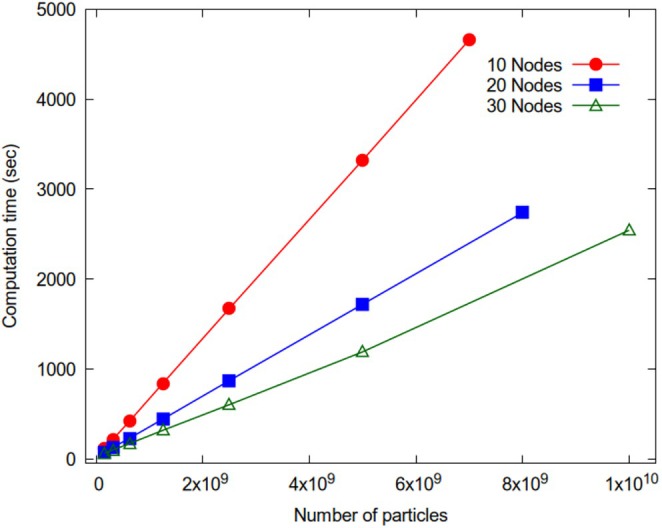
A parallelization strategy for hybrid particle-field molecular dynamics (hPF-MD) simulations on multi-node multi-GPU architectures is proposed. Two design principles have been followed to achieve a massively parallel version of the OCCAM code for distributed GPU computing: performing all the computations only on GPUs, minimizing data exchange between CPU and GPUs, and among GPUs. The hPF-MD scheme is particularly suitable to develop a GPU-resident and low data exchange code. Comparison of performances obtained using the previous multi-CPU code with the proposed multi-node multi-GPU version are reported. Several non-trivial issues to enable applications for systems of considerable sizes, including large input files handling and memory occupation, have been addressed. Large-scale benchmarks of hPF-MD simulations for system sizes up to 10 billion particles are presented. Performances obtained using a moderate quantity of computational resources highlight the feasibility of hPF-MD simulations in systematic studies of large-scale multibillion particle systems. This opens the possibility to perform systematic/routine studies and to reveal new molecular insights for problems on scales previously inaccessible to molecular simulations.
Keywords: GPU‐accelerated molecular dynamics; coarse‐graining; hybrid particle‐field method; large‐scale simulations.
© 2025 The Author(s). Journal of Computational Chemistry published by Wiley Periodicals LLC.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures












References
-
- De Nicola A., Touloupidis V., Kanellopoulos V., Albunia A. R., and Milano G., “A Combined Experimental and Molecular Simulation Study on Stress Generation Phenomena During the Ziegler‐Natta Polyethylene Catalyst Fragmentation Process,” Nanoscale Advances 4 (2022): 5178–5188, 10.1039/d2na00406b. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
