

Technical Evaluation of Plasma Proteomics Technologies
- PMID: 40366296
- PMCID: PMC12150332
- DOI: 10.1021/acs.jproteome.5c00221
Technical Evaluation of Plasma Proteomics Technologies
Abstract
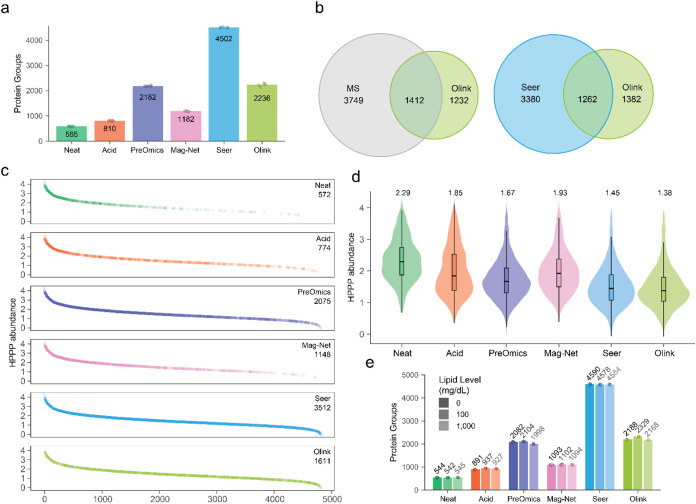
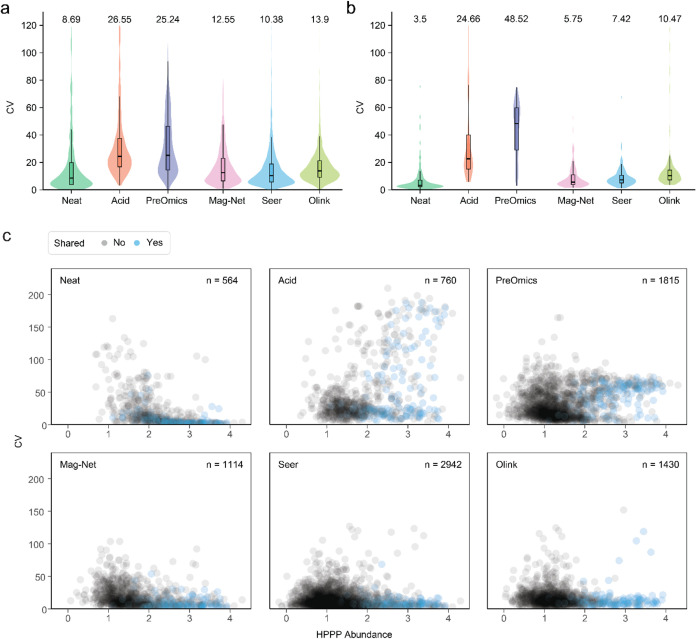
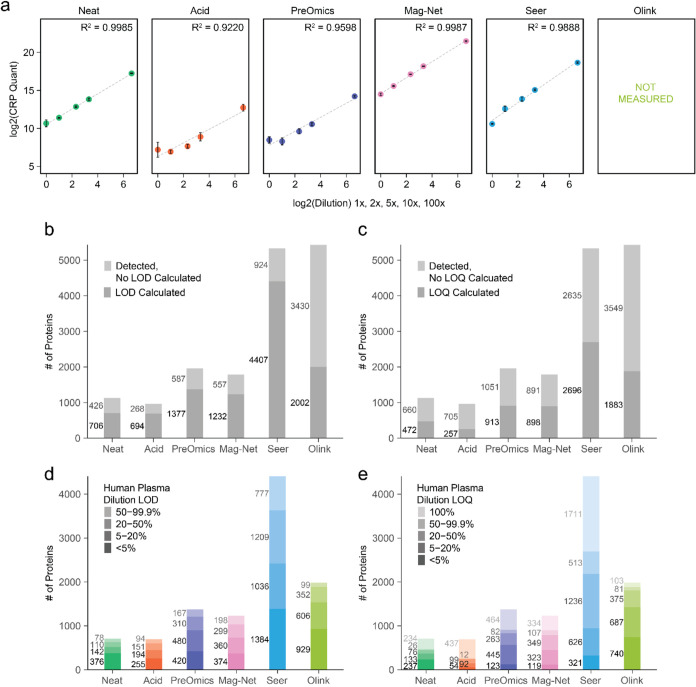
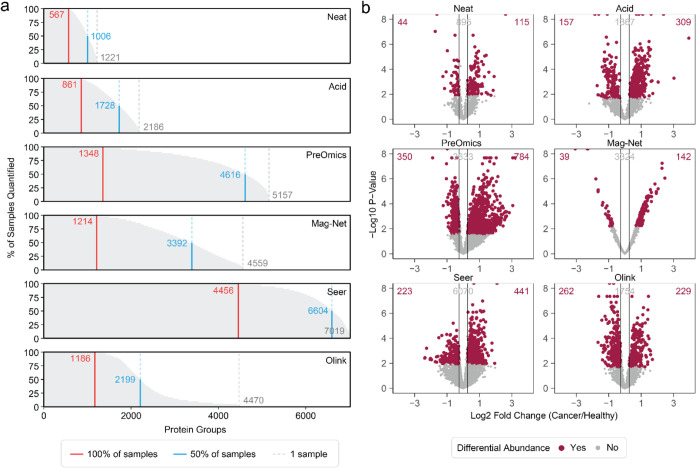
Plasma proteomics technologies are rapidly evolving and of critical importance to the field of biomedical research. Here, we report a technical evaluation of six notable plasma proteomics technologies─unenriched (Neat), acid depletion, PreOmics ENRICHplus, Mag-Net, Seer Proteograph XT, and Olink Explore HT. The methods were compared on proteomic depth, reproducibility, linearity, tolerance to lipid interference, and limit of detection/quantification. In total, we performed 618 LC-MS/MS experiments and 93 Olink Explore HT assays. The Seer method achieved the greatest proteomic depth (∼4500 proteins detected), while Olink detected ∼2600 proteins. Other MS-based methods ranged from ∼500-2200 proteins detected. In our analysis, Neat, Mag-Net, Seer, and Olink had good reproducibility, while PreOmics and Acid had higher variability (>20% median coefficient of variation). All MS methods showed good linearity with spiked-in C-reactive protein (CRP); CRP was surprisingly not in the Olink assay. None of the methods were affected by lipid interference. Seer produced the highest number of quantifiable proteins with a measurable LOD (4407) and LOQ (2696). Olink had the next highest number of quantifiable proteins, with 2002 having an LOD and 1883 having an LOQ. Finally, we tested the applicability of these methods for detecting differences between healthy and cancer groups in a nonsmall cell lung cancer (NSCLC) cohort. All six methods detected differentially abundant proteins between the cancer and healthy samples but disagreed on which proteins were significant, highlighting the contrast between each method.
Keywords: LC-MS; Mag-Net; Olink; PreOmics; Seer; mass spectrometry; method comparison; plasma; proteomics.
Figures
Update of
-
A Technical Evaluation of Plasma Proteomics Technologies.bioRxiv [Preprint]. 2025 Jan 13:2025.01.08.632035. doi: 10.1101/2025.01.08.632035. bioRxiv. 2025. Update in: J Proteome Res. 2025 Jun 6;24(6):3074-3087. doi: 10.1021/acs.jproteome.5c00221. PMID: 39868270 Free PMC article. Updated. Preprint.
References
-
- Larkin J. R., Anthony S., Johanssen V. A., Yeo T., Sealey M., Yates A. G., Smith C. F., Claridge T. D. W., Nicholson B. D., Moreland J.-A., Gleeson F., Sibson N. R., Anthony D. C., Probert F.. Metabolomic Biomarkers in Blood Samples Identify Cancers in a Mixed Population of Patients with Nonspecific Symptoms. Clin. Cancer Res. 2022;28(8):1651–1661. doi: 10.1158/1078-0432.CCR-21-2855. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
