

Divergent DNA methylation dynamics in marsupial and eutherian embryos
- PMID: 40369084
- PMCID: PMC12221971
- DOI: 10.1038/s41586-025-08992-2
Divergent DNA methylation dynamics in marsupial and eutherian embryos
Abstract
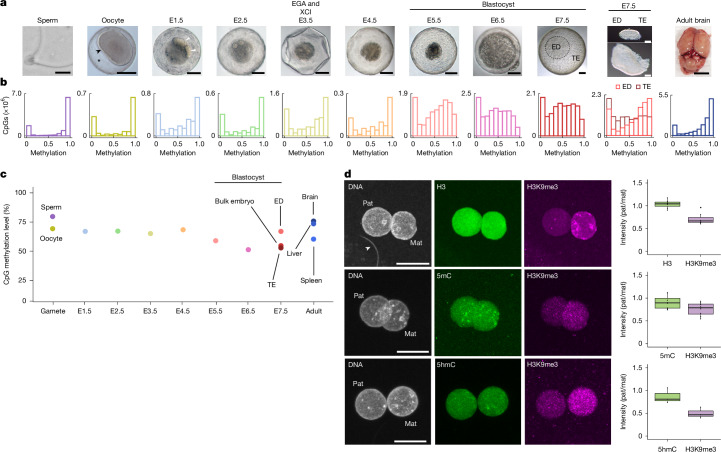
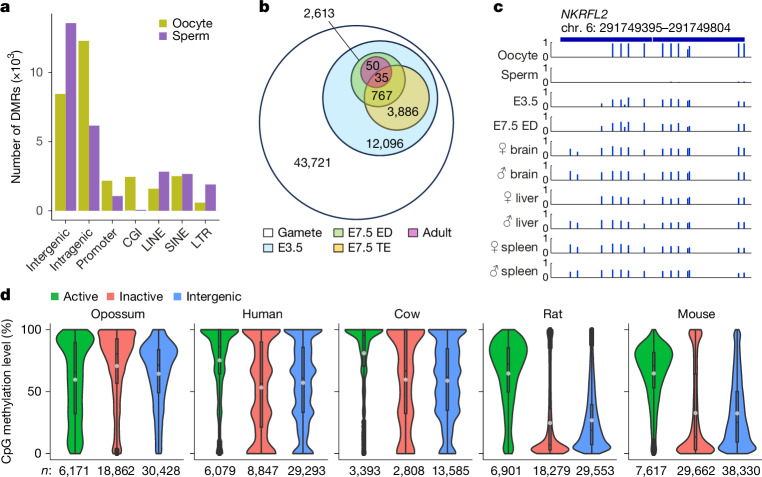
Based on seminal work in placental species (eutherians)1-10, a paradigm of mammalian development has emerged wherein the genome-wide erasure of parental DNA methylation is required for embryogenesis. Whether such DNA methylation reprogramming is, in fact, conserved in other mammals is unknown. Here, to resolve this point, we generated base-resolution DNA methylation maps in gametes, embryos and adult tissues of a marsupial, the opossum Monodelphis domestica, revealing variations from the eutherian-derived model. The difference in DNA methylation level between oocytes and sperm is less pronounced than that in eutherians. Furthermore, unlike the genome of eutherians, that of the opossum remains hypermethylated during the cleavage stages. In the blastocyst, DNA demethylation is transient and modest in the epiblast. However, it is sustained in the trophectoderm, suggesting an evolutionarily conserved function for DNA hypomethylation in the mammalian placenta. Furthermore, unlike that in eutherians, the inactive X chromosome becomes globally DNA hypomethylated during embryogenesis. We identify gamete differentially methylated regions that exhibit distinct fates in the embryo, with some transient, and others retained and that represent candidate imprinted loci. We also reveal a possible mechanism for imprinted X inactivation, through maternal DNA methylation of the Xist-like noncoding RNA RSX11. We conclude that the evolutionarily divergent eutherians and marsupials use DNA demethylation differently during embryogenesis.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures








References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
