

Evolutionary and immune microenvironment dynamics during neoadjuvant treatment of esophageal adenocarcinoma
- PMID: 40369175
- PMCID: PMC12122370
- DOI: 10.1038/s43018-025-00955-w
Evolutionary and immune microenvironment dynamics during neoadjuvant treatment of esophageal adenocarcinoma
Abstract
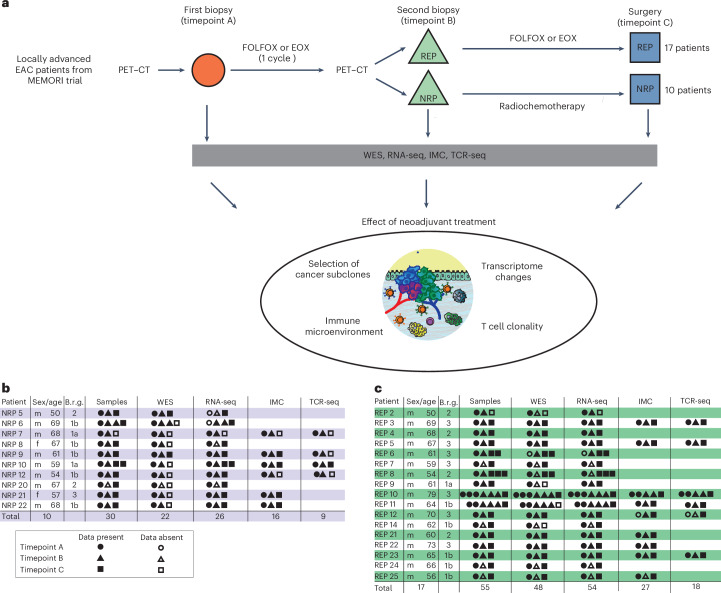
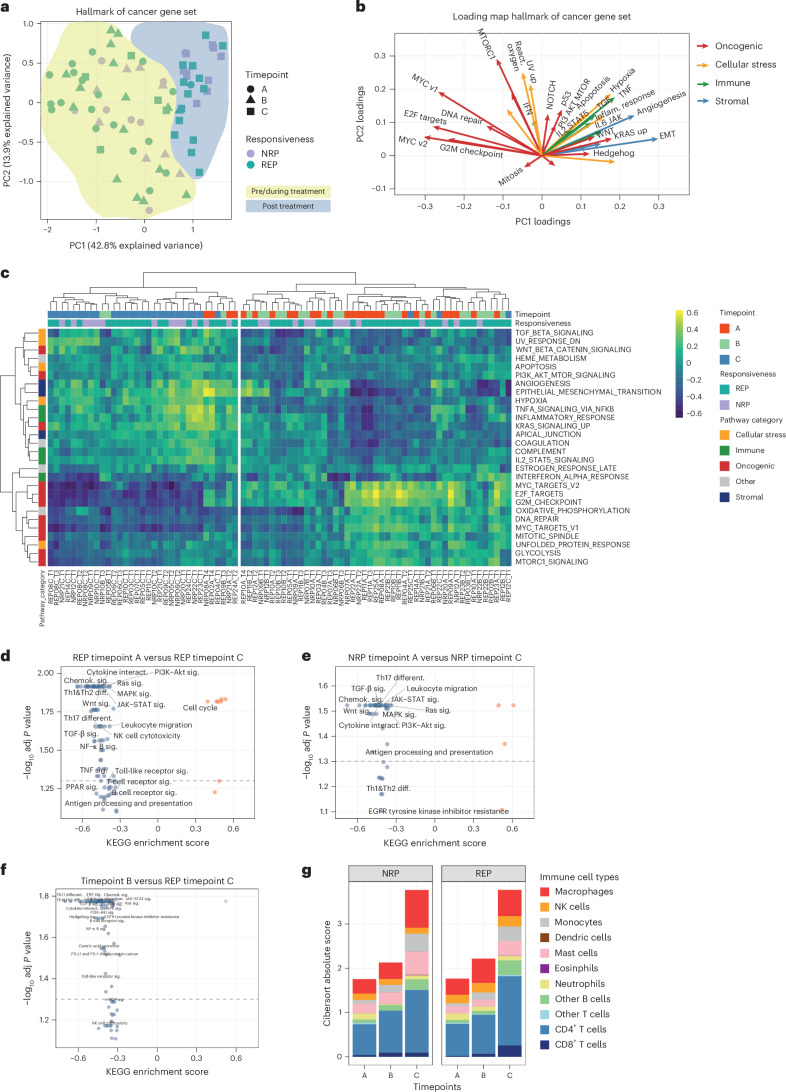
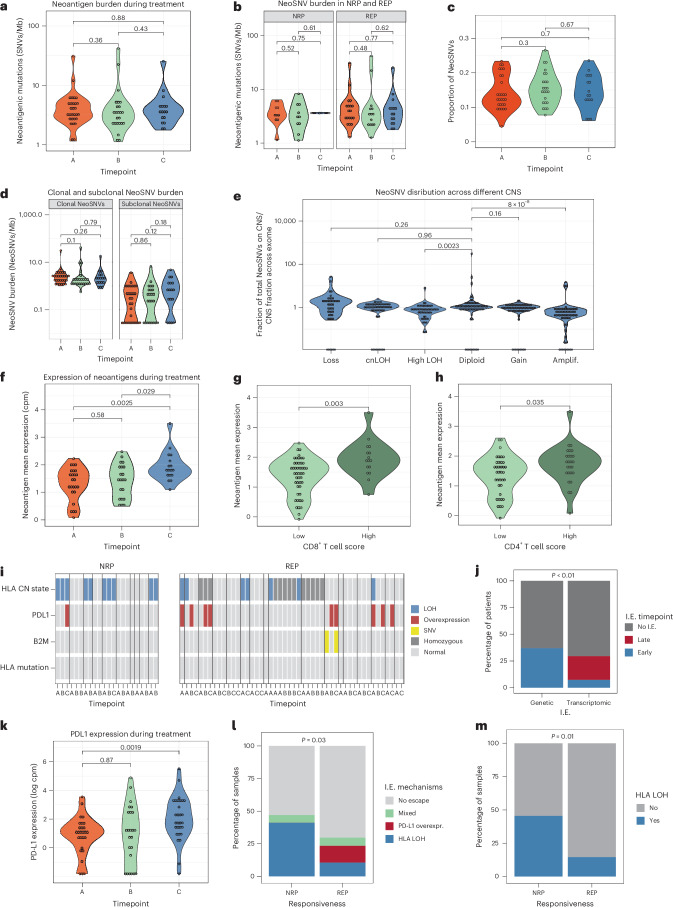
Locally advanced esophageal adenocarcinoma remains difficult to treat and the ecological and evolutionary dynamics responsible for resistance and recurrence are incompletely understood. Here, we performed longitudinal multiomic analysis of patients with esophageal adenocarcinoma in the MEMORI trial. Multi-region multi-timepoint whole-exome and paired transcriptome sequencing was performed on 27 patients before, during and after neoadjuvant treatment. We found major transcriptomic changes during treatment with upregulation of immune, stromal and oncogenic pathways. Genetic data revealed that clonal sweeps through treatment were rare. Imaging mass cytometry and T cell receptor sequencing revealed remodeling of the tumor microenvironment during treatment. The presence of genetic immune escape, a less-cytotoxic T cell phenotype and a lack of clonal T cell expansions were linked to poor treatment response. In summary, there were widespread transcriptional and environmental changes through treatment, with limited clonal replacement, suggestive of phenotypic plasticity.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: J.T.S. receives honoraria as a consultant or for continuing medical education presentations from AstraZeneca, Bayer, Boehringer Ingelheim, Bristol Myers Squibb, Immunocore, iOMEDICO, MSD, Novartis, Roche/Genentech and Servier. His institution receives research funding from Abalos Therapeutics, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Eisbach Bio and Roche/Genentech; he holds ownership in FAPI Holding. K. Steiger is named on a patent on a radiopharmaceutical compound and serves as advisory board member for TRIMT GmbH; not related to the current work. M.Q. receives honoraria as consultant or for continuing medical education presentations from AstraZeneca, Bayer, Bristol-Myers Squibb, MSD Sharp Dohme, Novartis, Roche and Servier. T.A.G., B.C. and A.M.B. are named as co-inventors on patent applications that describe a method for TCR sequencing (GB2305655.9) and T.A.G. is named on a patent application for a method to measure evolutionary dynamics in cancers using DNA methylation (GB2317139.0). T.A.G. has received an honorarium from Genentech and consultancy fees from DAiNA Therapeutics. The remaining authors declare no competing interests.
Figures














Update of
-
Evolutionary and immune microenvironment dynamics during neoadjuvant treatment of oesophagael adenocarcinoma.Res Sq [Preprint]. 2023 Apr 13:rs.3.rs-2738048. doi: 10.21203/rs.3.rs-2738048/v1. Res Sq. 2023. Update in: Nat Cancer. 2025 May;6(5):820-837. doi: 10.1038/s43018-025-00955-w. PMID: 37090678 Free PMC article. Updated. Preprint.
References
-
- Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin.10.3322/caac.21660 (2021). - PubMed
-
- Morgan, E. et al. International trends in oesophageal cancer survival by histological subtype between 1995 and 2014. Gut70, 234–242 (2021). - PubMed
-
- Davies, A. R. et al. Tumor stage after neoadjuvant chemotherapy determines survival after surgery for adenocarcinoma of the esophagus and esophagogastric junction. J. Clin. Oncol.32, 2983–2990 (2014). - PubMed
-
- Al-Batran, S. E. et al. Perioperative chemotherapy with fluorouracil plus leucovorin, oxaliplatin, and docetaxel versus fluorouracil or capecitabine plus cisplatin and epirubicin for locally advanced, resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4): a randomised, phase 2/3 trial. Lancet393, 1948–1957 (2019). - PubMed
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
