

Type 1 Diabetes: A Guide to Autoimmune Mechanisms for Clinicians
- PMID: 40375390
- PMCID: PMC12312826
- DOI: 10.1111/dom.16460
Type 1 Diabetes: A Guide to Autoimmune Mechanisms for Clinicians
Abstract
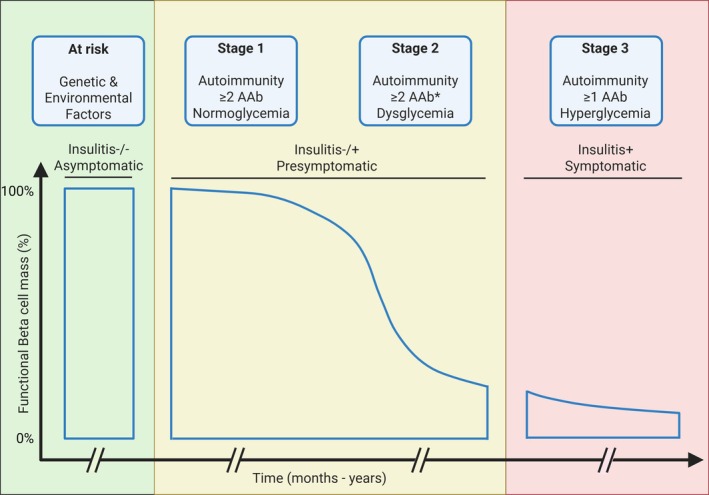
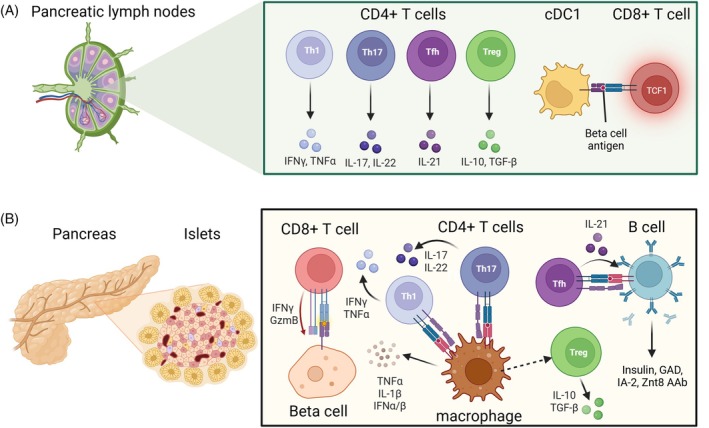
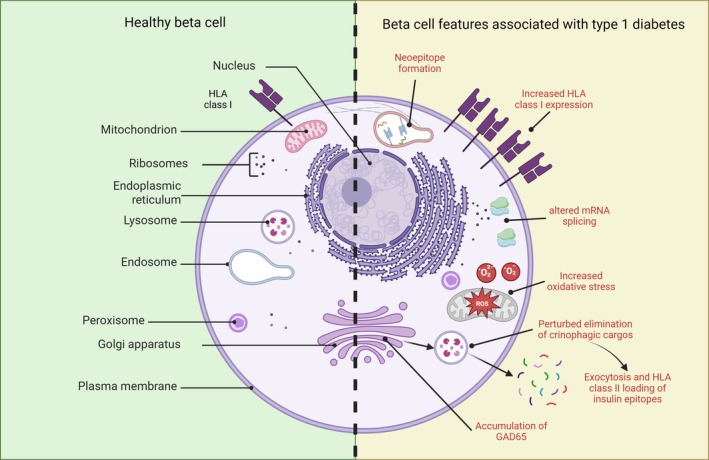
Type 1 diabetes (T1D) results from the destruction of pancreatic beta cells by autoreactive T lymphocytes, leading to insulin deficiency and lifelong insulin dependence. It develops in genetically predisposed individuals, triggered by environmental or immunological factors. Although the exact causes of T1D remain unknown, the autoimmune pathogenesis of the disease is clearly indicated by the genetic risk conferred by allelic human leukocyte antigens (HLA), the almost obligatory presence of islet cell autoantibodies (AAbs) and immune cell infiltration of pancreatic islets from patients. At the same time, epidemiological data point to a role of environmental factors, notably enteroviral infections, in the disease, although precise causative links between specific pathogens and T1D have been difficult to establish. Studies of human pancreas organs from patients made available through repositories and the advent of high-dimensional high-throughput technologies for genomic and proteomic studies have significantly elucidated our understanding of the disease in recent years and provided mechanistic insights that can be exploited for innovative targeted therapeutic approaches. This short overview will summarise current salient knowledge on immune cell and beta cell dysfunction in T1D pathogenesis. PLAIN LANGUAGE SUMMARY: Type 1 diabetes (T1D) is a chronic disease where the body's own immune system attacks and destroys the insulin-producing beta cells in the pancreas. This leads to a lack of insulin, a hormone essential for regulating blood sugar, which means people with T1D need insulin for life. The disease can develop at any age but is most diagnosed in children and young adults. Despite advances in treatment, T1D still significantly reduces life expectancy, especially in countries with fewer healthcare resources. T1D develops in people with a genetic predisposition, often triggered by environmental factors such as viral infections or changes in the gut microbiome. The disease progresses silently through three stages: Stage 1: Autoantibodies to beta cell components appear, signalling the immune system is reacting against the pancreas, but there are no symptoms; Stage 2: Beta cell function starts to decline, but fasting blood sugar is still normal; Stage 3: Enough beta cells are destroyed that fasting blood sugar rises, and symptoms of diabetes appear. The risk of progressing from stage 1 to full-blown diabetes is about 35-50% within five years, and even higher from stage 2. Over 60 genes are linked to T1D risk, most of which affect how the immune system works. The strongest genetic risk comes from specific versions of histocompatibility genes, which help the immune system distinguish between the body's own cells and invaders. Some types of these genes make it easier for the immune system to mistakenly attack beta cells. However, 90% of people diagnosed with T1D have no family member with T1D, showing that genetics is only part of the story. Environmental factors also play a big role. For example, certain viral infections, especially with viruses infecting the intestine, are associated with a higher risk of developing T1D. The gut microbiome - the community of bacteria living in our intestines - also influences risk, with healthier, more diverse microbiomes appearing to offer some protection. In T1D, immune cells - especially so-called T lymphocytes - mistake beta cells in the pancreas for threats and destroy them. This process is called autoimmunity. The attack is often reflected by the presence of autoantibodies against proteins found in beta cells. Over time, as more beta cells are lost, the body can no longer produce enough insulin, leading to the symptoms of diabetes. Interestingly, not all people with T1D have the same pattern of disease. For example, children diagnosed before age 7 often have more aggressive disease, more autoantibodies, and stronger genetic risk factors than those diagnosed later. Much of our understanding of T1D has come from studying animal models, but new technologies now allow researchers to study human pancreas tissue and blood immune cells in greater detail. Scientists are also exploring how the gut microbiome, diet, and environmental exposures contribute to T1D risk and progression. Treatment currently focuses on replacing insulin, but researchers are working on therapies that target the immune system or aim to protect or replace beta cells. Strategies include immunotherapy, gene therapy, and even modifying the gut microbiome. The goal is to prevent or reverse the disease, not just manage its symptoms. In summary, T1D is a complex autoimmune disease influenced by both genes and the environment. It progresses silently before symptoms appear, and while insulin therapy is life-saving, new research is paving the way for treatments that could one day halt or even prevent the disease.
Keywords: cellular research; islets; mouse model; type 1 diabetes.
© 2025 The Author(s). Diabetes, Obesity and Metabolism published by John Wiley & Sons Ltd.
Conflict of interest statement
P.M.v.E. and F.‐X.M. declared enough conflict of interest.
Figures
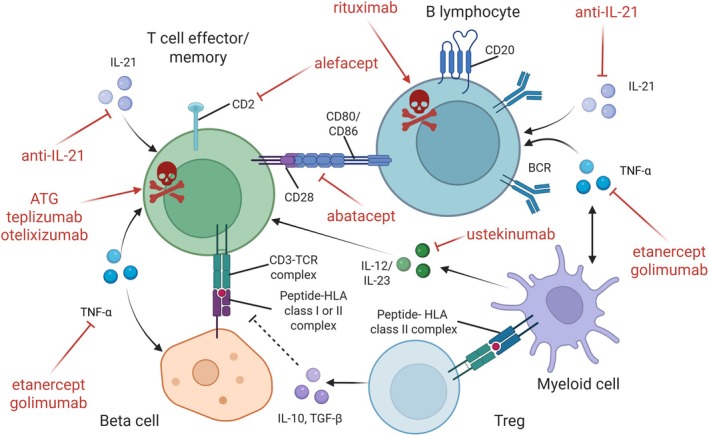
 ) through antibodies to CD3, CD2 or CD20, blocking co‐stimulation through inhibition of CD28‐CD80/86 interaction, or inhibiting cytokines such as IL‐21, IL‐12, IL‐23 and TNF‐α. The illustration also mentions cell therapy by polyclonal Tregs although the benefit has been modest so far. Approaches targeting intracellular signalling by kinases are not displayed, as are autoantigen‐based strategies. See Table 1 for details and references.
) through antibodies to CD3, CD2 or CD20, blocking co‐stimulation through inhibition of CD28‐CD80/86 interaction, or inhibiting cytokines such as IL‐21, IL‐12, IL‐23 and TNF‐α. The illustration also mentions cell therapy by polyclonal Tregs although the benefit has been modest so far. Approaches targeting intracellular signalling by kinases are not displayed, as are autoantigen‐based strategies. See Table 1 for details and references.Similar articles
-
The extra-islet pancreas supports autoimmunity in human type 1 diabetes.Elife. 2025 Apr 15;13:RP100535. doi: 10.7554/eLife.100535. Elife. 2025. PMID: 40232951 Free PMC article.
-
The Changing Epidemiology of Type 1 Diabetes: A Global Perspective.Diabetes Obes Metab. 2025 Aug;27 Suppl 6(Suppl 6):3-14. doi: 10.1111/dom.16501. Epub 2025 Jun 19. Diabetes Obes Metab. 2025. PMID: 40536127 Free PMC article. Review.
-
The Black Book of Psychotropic Dosing and Monitoring.Psychopharmacol Bull. 2024 Jul 8;54(3):8-59. Psychopharmacol Bull. 2024. PMID: 38993656 Free PMC article. Review.
-
Health economic considerations of screening for early type 1 diabetes.Diabetes Obes Metab. 2025 Aug;27 Suppl 6(Suppl 6):69-77. doi: 10.1111/dom.16522. Epub 2025 Jun 24. Diabetes Obes Metab. 2025. PMID: 40555704 Free PMC article. Review.
-
Sexual Harassment and Prevention Training.2024 Mar 29. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Mar 29. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 36508513 Free Books & Documents.
References
-
- Gregory GA, Robinson TIG, Linklater SE, et al. Global incidence, prevalence, and mortality of type 1 diabetes in 2021 with projection to 2040: a modelling study. Lancet Diabetes Endocrinol. 2022;10:741‐747. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
