

Similarity-weighted entropy for quantifying genetic diversity in viral quasispecies
- PMID: 40376429
- PMCID: PMC12079039
- DOI: 10.1093/ve/veaf029
Similarity-weighted entropy for quantifying genetic diversity in viral quasispecies
Abstract
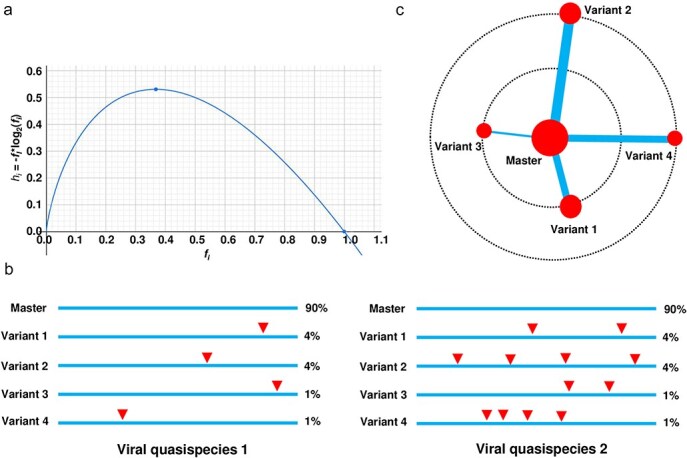
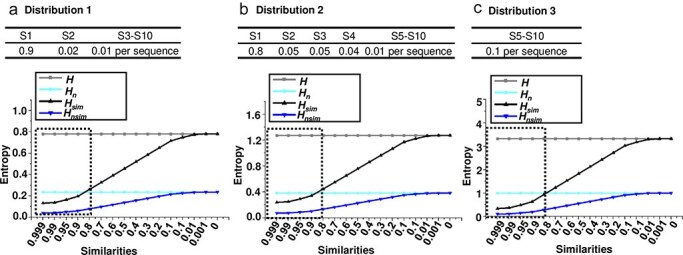
A viral quasispecies is a genetically diverse population of closely related viral variants that exist in a state of dynamic equilibrium. This diversity, driven by mutations, recombination, and selective pressures, enables viruses to adapt rapidly, affecting pathogenicity and treatment resistance. Quantifying the genetic diversity within viral quasispecies is therefore crucial for understanding viral evolution and for designing effective therapeutic strategies. Entropy is a commonly used metric to measure genetic diversity within such populations; however, traditional entropy calculations often neglect genetic similarities between sequences, which can result in overestimating true diversity. In this study, I compare several widely used diversity indices for quantifying viral quasispecies diversity and introduce a novel similarity-weighted entropy metric that incorporates sequence similarity into entropy calculations. This approach enables a more comprehensive representation of diversity in genetically cohesive viral populations. By applying both conventional and similarity-weighted entropy calculations to hypothetical sequence populations and real viroid and virus quasispecies, I demonstrate that similarity-weighted entropy provides a more comprehensive measure of genetic diversity while maintaining the simplicity of conventional entropy. These findings highlight the value of similarity-weighted entropy in characterizing viral quasispecies and its potential to improve our understanding of viral adaptation and resistance mechanisms.
Keywords: Viral quasispecies; entropy; genetic diversity; sequence similarity.
© The Author(s) 2025. Published by Oxford University Press.
Conflict of interest statement
None declared.
Figures




Similar articles
-
aBayesQR: A Bayesian Method for Reconstruction of Viral Populations Characterized by Low Diversity.J Comput Biol. 2018 Jul;25(7):637-648. doi: 10.1089/cmb.2017.0249. Epub 2018 Feb 26. J Comput Biol. 2018. PMID: 29480740
-
Similarities between Human Immunodeficiency Virus Type 1 and Hepatitis C Virus Genetic and Phenotypic Protease Quasispecies Diversity.J Virol. 2015 Oct;89(19):9758-64. doi: 10.1128/JVI.01097-15. Epub 2015 Jul 15. J Virol. 2015. PMID: 26178979 Free PMC article.
-
Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population.Nature. 2006 Jan 19;439(7074):344-8. doi: 10.1038/nature04388. Epub 2005 Dec 4. Nature. 2006. PMID: 16327776 Free PMC article.
-
Quasispecies theory and the behavior of RNA viruses.PLoS Pathog. 2010 Jul 22;6(7):e1001005. doi: 10.1371/journal.ppat.1001005. PLoS Pathog. 2010. PMID: 20661479 Free PMC article. Review.
-
Historical Perspective on the Discovery of the Quasispecies Concept.Annu Rev Virol. 2021 Sep 29;8(1):51-72. doi: 10.1146/annurev-virology-091919-105900. Annu Rev Virol. 2021. PMID: 34586874 Review.
References
LinkOut - more resources
Full Text Sources
