

Auricular Transcutaneous Vagus Nerve Stimulation Enhances Post-Stroke Neurological and Cognitive Recovery in Mice by Suppressing Ferroptosis Through α7 Nicotinic Acetylcholine Receptor Activation
- PMID: 40376919
- PMCID: PMC12082278
- DOI: 10.1111/cns.70439
Auricular Transcutaneous Vagus Nerve Stimulation Enhances Post-Stroke Neurological and Cognitive Recovery in Mice by Suppressing Ferroptosis Through α7 Nicotinic Acetylcholine Receptor Activation
Abstract
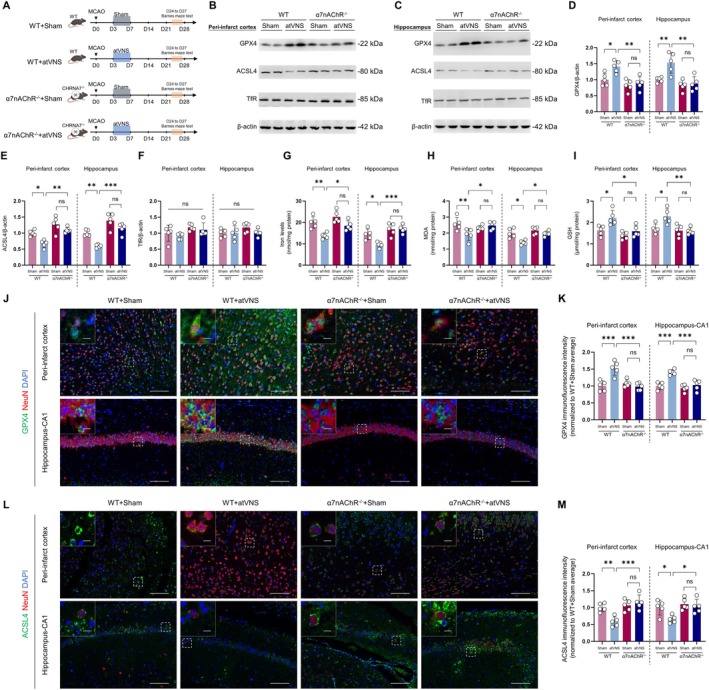
Aims: Ferroptosis plays a critical role in stroke pathophysiology, yet its dynamics during recovery remain unclear. This study aimed to investigate the evolution of ferroptosis throughout post-stroke recovery and evaluate auricular transcutaneous vagus nerve stimulation (atVNS) as a therapeutic intervention, focusing on the involvement of α7 nicotinic acetylcholine receptor (α7nAChR)-mediated mechanisms.
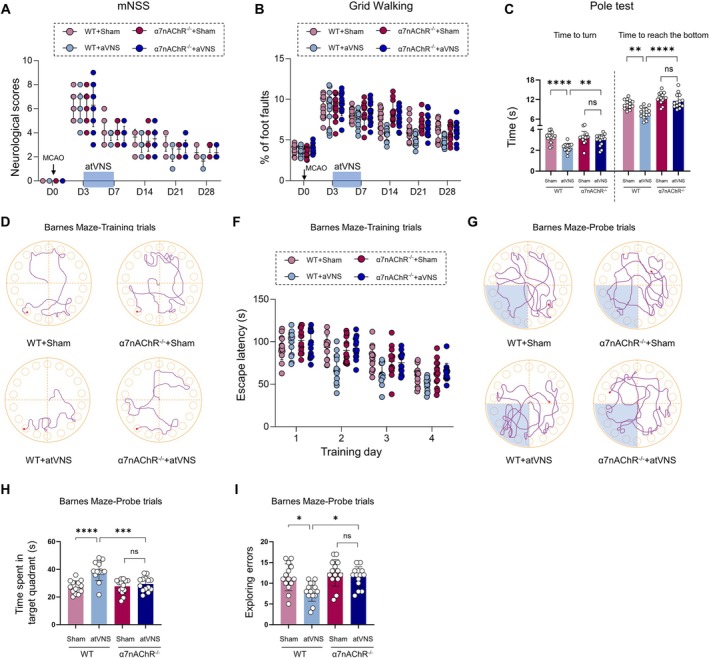
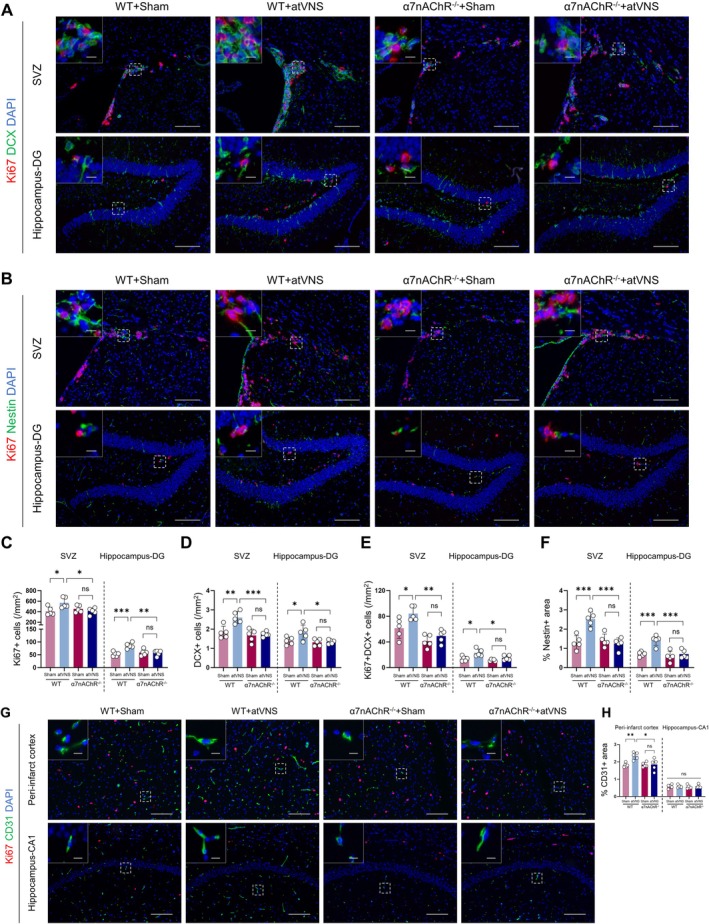
Methods: Using a middle cerebral artery occlusion (MCAO) mouse model, we examined ferroptosis-related protein expression (GPX4, ACSL4, TfR) and iron levels across acute to chronic recovery phases. The therapeutic effects of atVNS were evaluated through the assessment of ferroptosis markers, neurogenesis, angiogenesis, cognitive function, and neuroinflammation. α7nAChR knockout mice were used to investigate the receptor's role in atVNS-mediated recovery.
Results: We observed sustained alterations in ferroptosis markers and iron levels throughout post-stroke recovery. atVNS treatment reduced ferroptosis progression by modulating GPX4 and ACSL4 expression, enhanced neurogenesis and angiogenesis, improved cognitive recovery, and reduced neuroinflammation. These beneficial effects were absent in α7nAChR knockout mice, while atVNS increased neuronal α7nAChR expression in wild-type mice.
Conclusions: This study reveals the persistent involvement of ferroptosis in stroke recovery and demonstrates that atVNS provides comprehensive neuroprotection through α7nAChR-dependent mechanisms. These findings establish atVNS as a promising noninvasive therapeutic approach for stroke recovery and highlight α7nAChR signaling as a potential therapeutic target.
Keywords: auricular transcutaneous vagus nerve stimulation; ferroptosis; neurogenesis; neuroinflammation; α7 nicotinic acetylcholine receptor.
© 2025 The Author(s). CNS Neuroscience & Therapeutics published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures








Similar articles
-
Vagus nerve stimulation as a promising neuroprotection for ischemic stroke via α7nAchR-dependent inactivation of microglial NLRP3 inflammasome.Acta Pharmacol Sin. 2024 Jul;45(7):1349-1365. doi: 10.1038/s41401-024-01245-4. Epub 2024 Mar 19. Acta Pharmacol Sin. 2024. PMID: 38504011 Free PMC article.
-
α7nAchR mediates transcutaneous auricular vagus nerve stimulation-induced neuroprotection in a rat model of ischemic stroke by enhancing axonal plasticity.Neurosci Lett. 2020 Jun 21;730:135031. doi: 10.1016/j.neulet.2020.135031. Epub 2020 May 19. Neurosci Lett. 2020. PMID: 32416113
-
Mechanisms underlying antidepressant effect of transcutaneous auricular vagus nerve stimulation on CUMS model rats based on hippocampal α7nAchR/NF-κB signal pathway.J Neuroinflammation. 2021 Dec 17;18(1):291. doi: 10.1186/s12974-021-02341-6. J Neuroinflammation. 2021. PMID: 34920740 Free PMC article.
-
Vagus nerve stimulation: a new bioelectronics approach to treat rheumatoid arthritis?Best Pract Res Clin Rheumatol. 2014 Aug;28(4):625-35. doi: 10.1016/j.berh.2014.10.015. Epub 2014 Nov 20. Best Pract Res Clin Rheumatol. 2014. PMID: 25481554 Review.
-
New Insight into Neuropathic Pain: The Relationship between α7nAChR, Ferroptosis, and Neuroinflammation.Int J Mol Sci. 2024 Jun 18;25(12):6716. doi: 10.3390/ijms25126716. Int J Mol Sci. 2024. PMID: 38928421 Free PMC article. Review.
References
-
- Vázquez‐Oliver A., Brambilla‐Pisoni C., Domingo‐Gainza M., Maldonado R., Ivorra A., and Ozaita A., “Auricular Transcutaneous Vagus Nerve Stimulation Improves Memory Persistence in Naïve Mice and in an Intellectual Disability Mouse Model,” Brain Stimulation 13, no. 2 (2020): 494–498. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
