

Unraveling immunosenescence in sepsis: from cellular mechanisms to therapeutics
- PMID: 40379629
- PMCID: PMC12084380
- DOI: 10.1038/s41419-025-07714-w
Unraveling immunosenescence in sepsis: from cellular mechanisms to therapeutics
Abstract
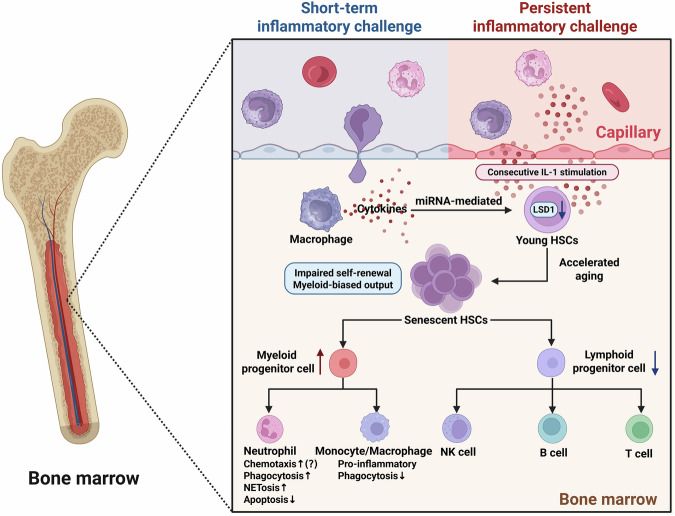
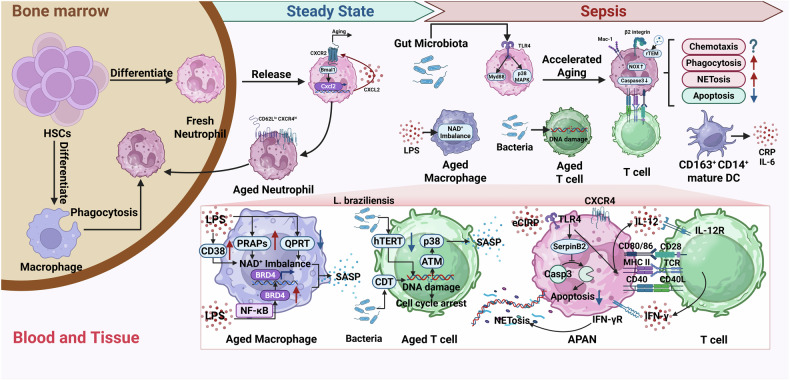
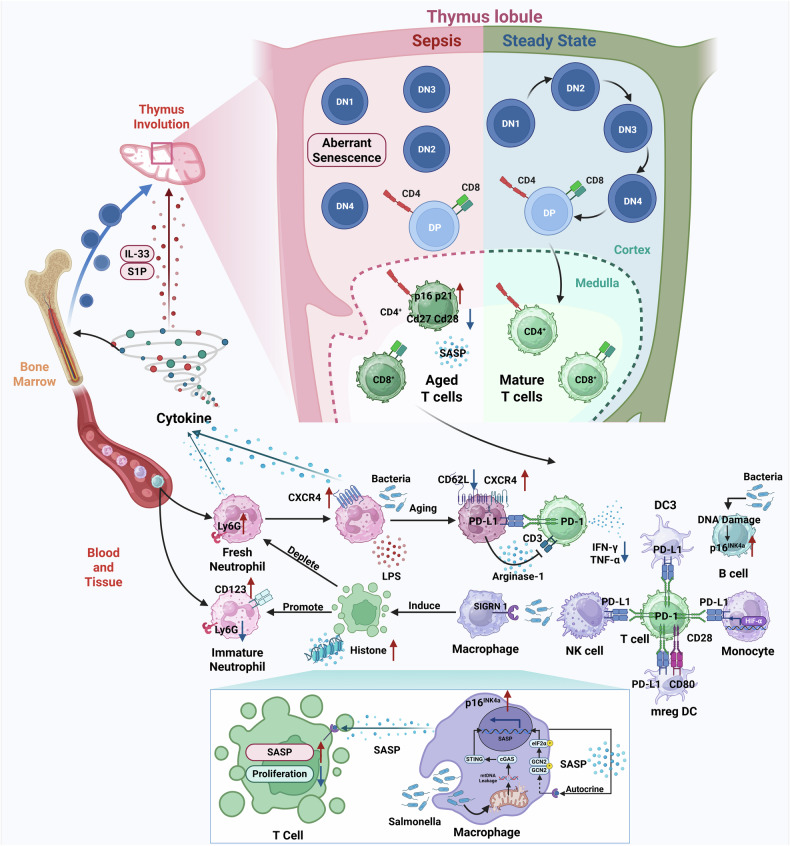
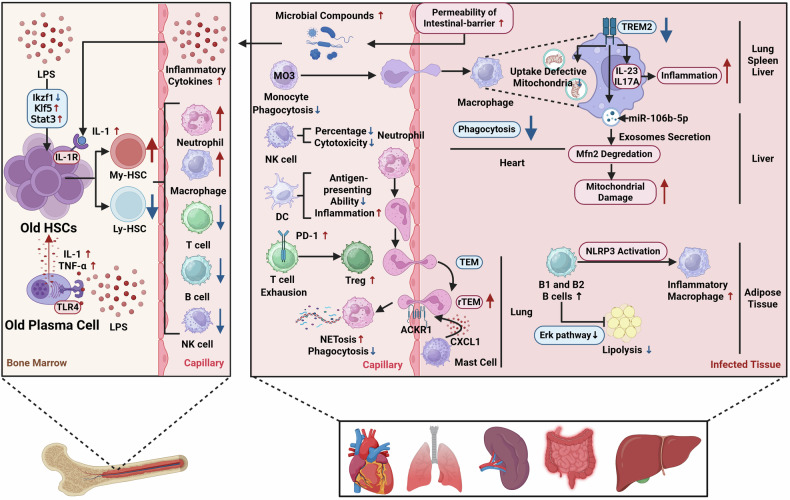
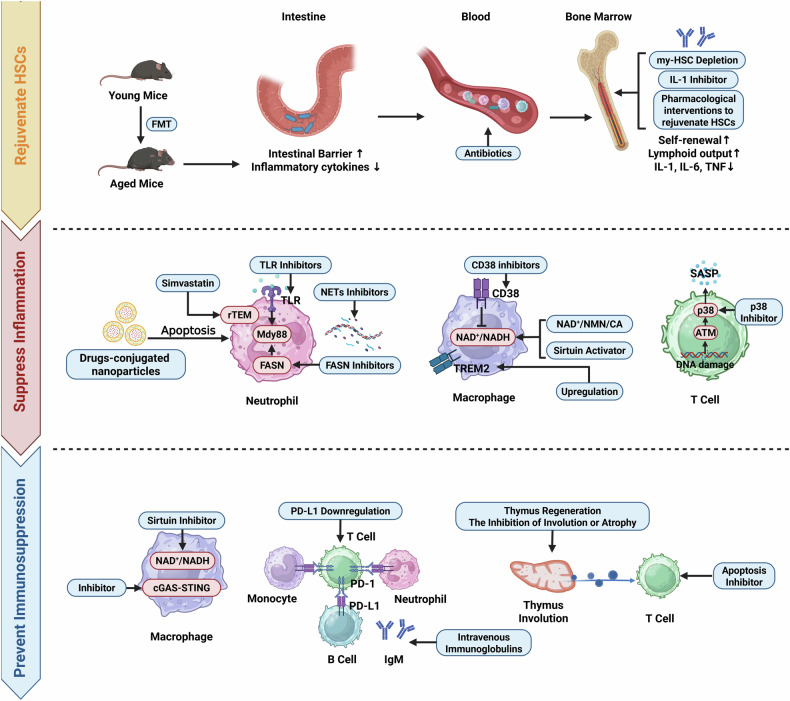
Sepsis is a life-threatening multiple organ dysfunction resulting from a dysregulated host response to infection, and patients with sepsis always exhibit a state of immune disorder characterized by both overwhelming inflammation and immunosuppression. The aging of immune system, namely "immunosenescence", has been reported to be correlated with high morbidity and mortality in elderly patients with sepsis. Initially, immunosenescence was considered as a range of age-related alterations in the immune system. However, increasing evidence has proven that persistent inflammation or even a short-term inflammatory challenge during sepsis could trigger accelerated aging of immune cells, which might further exacerbate inflammatory cytokine storm and promote the shift towards immunosuppression. Thus, premature immunosenescence is found in young sepsis individuals, which further aggravates immune disorders and induces the progression of sepsis. Furthermore, in old sepsis patients, the synergistic effects of both sepsis and aging may cause immunosenescence-associated alterations more significantly, resulting in more severe immune dysfunction and a worse prognosis. Therefore, it is necessary to explore the potential therapeutic strategies targeting immunosenescence during sepsis.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Immunosenescence and age-related immune cells: causes of age-related diseases.Arch Pharm Res. 2025 Feb;48(2):132-149. doi: 10.1007/s12272-024-01529-7. Epub 2024 Dec 26. Arch Pharm Res. 2025. PMID: 39725853 Review.
-
Immunosenescence: A Critical Factor Associated With Organ Injury After Sepsis.Front Immunol. 2022 Jul 18;13:917293. doi: 10.3389/fimmu.2022.917293. eCollection 2022. Front Immunol. 2022. PMID: 35924237 Free PMC article. Review.
-
Immunosenescence in Sepsis: Molecular Mechanisms and Potential Therapeutic Targets.Aging Dis. 2025 Mar 14. doi: 10.14336/AD.2025.0039. Online ahead of print. Aging Dis. 2025. PMID: 40153575 Review.
-
Cellular Senescence, Immunosenescence and HIV.Interdiscip Top Gerontol Geriatr. 2017;42:28-46. doi: 10.1159/000448542. Epub 2016 Nov 22. Interdiscip Top Gerontol Geriatr. 2017. PMID: 27875822 Review.
-
Recent Advances in Aging and Immunosenescence: Mechanisms and Therapeutic Strategies.Cells. 2025 Mar 27;14(7):499. doi: 10.3390/cells14070499. Cells. 2025. PMID: 40214453 Free PMC article. Review.
Cited by
-
Clinical Characterization, Risk Factors, and Mortality in Patients with Carbapenem-Resistant Hypervirulent Klebsiella pneumoniae Intra-Abdominal Infections.Infect Drug Resist. 2025 Jul 23;18:3647-3660. doi: 10.2147/IDR.S529532. eCollection 2025. Infect Drug Resist. 2025. PMID: 40718370 Free PMC article.
-
Frailty Index-laboratory and lymphocyte subset patterns in predicting 28-day mortality among elderly sepsis patients: a multicenter observational cohort study.Front Immunol. 2025 Jul 16;16:1624655. doi: 10.3389/fimmu.2025.1624655. eCollection 2025. Front Immunol. 2025. PMID: 40740787 Free PMC article.
References
-
- Cajander S, Kox M, Scicluna BP, Weigand MA, Mora RA, Flohé SB, et al. Profiling the dysregulated immune response in sepsis: overcoming challenges to achieve the goal of precision medicine. Lancet Respir Med. 2024;12:305–22. - PubMed
-
- Giamarellos-Bourboulis EJ, Aschenbrenner AC, Bauer M, Bock C, Calandra T, Gat-Viks I, et al. The pathophysiology of sepsis and precision-medicine-based immunotherapy. Nat Immunol. 2024;25:19–28. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
