

Comparison of RNA- and DNA-based 16S amplicon sequencing to find the optimal approach for the analysis of the uterine microbiome
- PMID: 40379732
- PMCID: PMC12084623
- DOI: 10.1038/s41598-025-00969-5
Comparison of RNA- and DNA-based 16S amplicon sequencing to find the optimal approach for the analysis of the uterine microbiome
Abstract
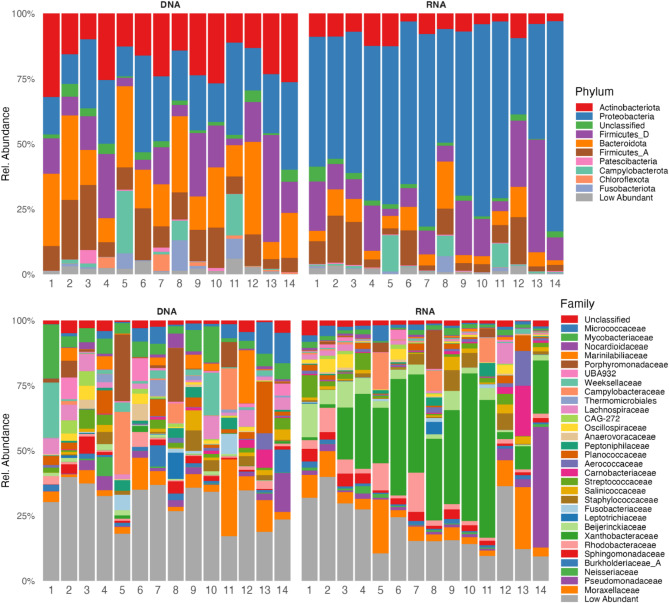
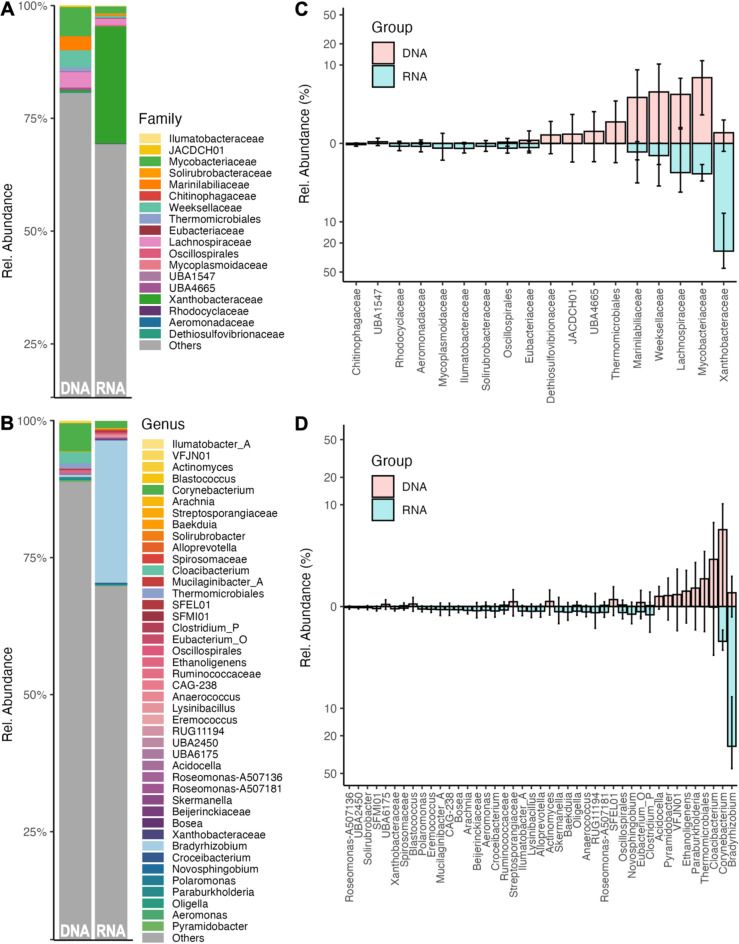
Studies in humans and large animals indicate a relationship between the uterine microbiome composition and endometrial receptivity. Despite many studies have been performed, the analysis of the uterine microbiome remains challenging due to the very low microbial biomass. Studies in other biological systems showed that RNA-based microbiome analysis complements DNA-based results and provides information about active bacteria in a sample. Thus, the aim of this study was to establish a highly sensitive and specific 16S rRNA gene V3-V4 amplicon PCR from equine uterine cytobrush samples and to compare DNA- and RNA-based 16S rRNA microbiome analysis. An optimized 16S rRNA gene V3-V4 amplicon PCR protocol from equine uterine cytobrush samples was developed, which was able to detect less than 38 bacterial genome copies using a bacterial DNA community standard. For the RNA-based amplicon generation protocol starting from cDNA, at least a 10-fold higher sensitivity was estimated compared to DNA-based approach. The comparison of using RNA and DNA isolated from the same uterine cytobrush samples as input for 16S V3-V4 amplicon sequencing revealed a much higher number of amplicon sequence variants as well as taxonomic units for the RNA-based approach. This resulted in significant differences in alpha (Simpson, Chao1) and beta diversity between RNA- and DNA-based analysis. Differential abundance analysis revealed significant differences between DNA and RNA samples at all taxonomic levels. Despite these differences, the overall microbiome composition was similar between the paired DNA and RNA samples. Many differences were probably found due to the higher sensitivity of the RNA-based approach. Furthermore, the DNA-based analysis is biased by the rRNA gene copy numbers (1-21), and the RNA-based analysis by the number of ribosomes per cell, which was reflected in the differences in the microbiome composition between the approaches. In addition, the results suggested that the DNA-based analysis is detecting cell-free bacterial DNA and/or DNA of dead bacteria that could be present in the samples. Altogether, the obtained results indicate advantages of a combined DNA- and RNA-based microbiome analysis, offering complementary and valuable information in the context of fertility-related studies of the uterine microbiome.
Keywords: Equus caballus; Bacterial viability; Endometrial health; Microbial community analysis; Reproductive pathology.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests. Ethics approval and consent to participate: The samples were collected from mares owned by the University of Illinois Urbana-Champaign or from client-owned mares presented at the Veterinary Hospital (IACUC ethical approvals 21237 and 21238, respectively).
Figures
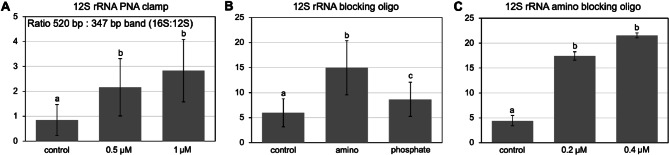
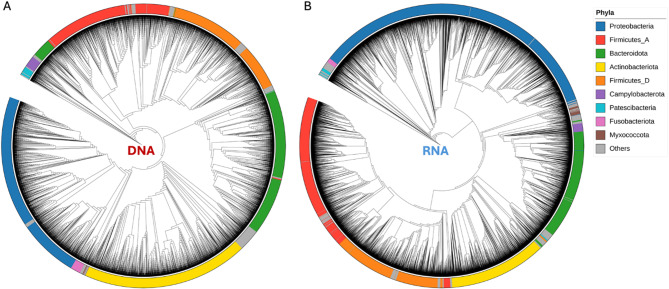
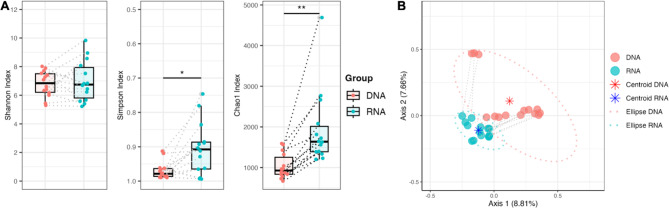
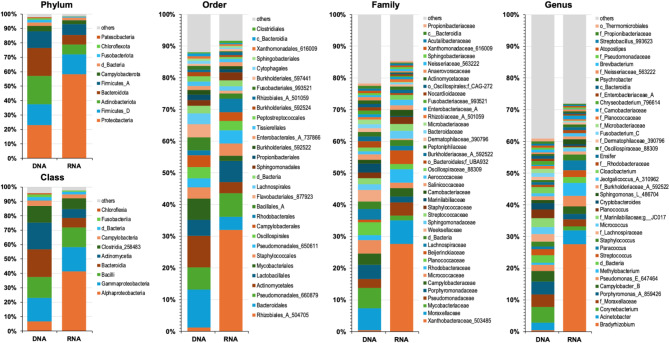
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
