

Repeatability of evolution and genomic predictions of temperature adaptation in seed beetles
- PMID: 40379980
- PMCID: PMC12148939
- DOI: 10.1038/s41559-025-02716-5
Repeatability of evolution and genomic predictions of temperature adaptation in seed beetles
Abstract
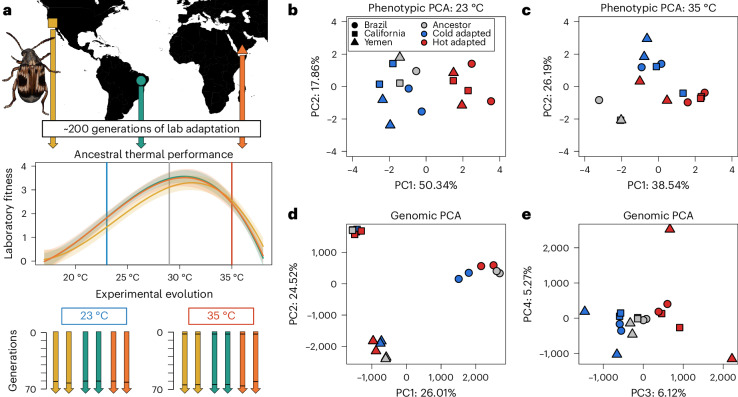
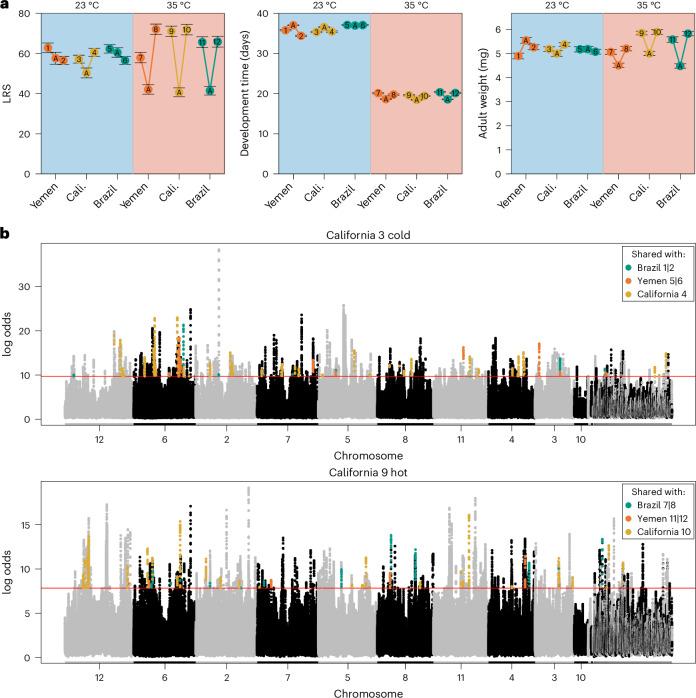
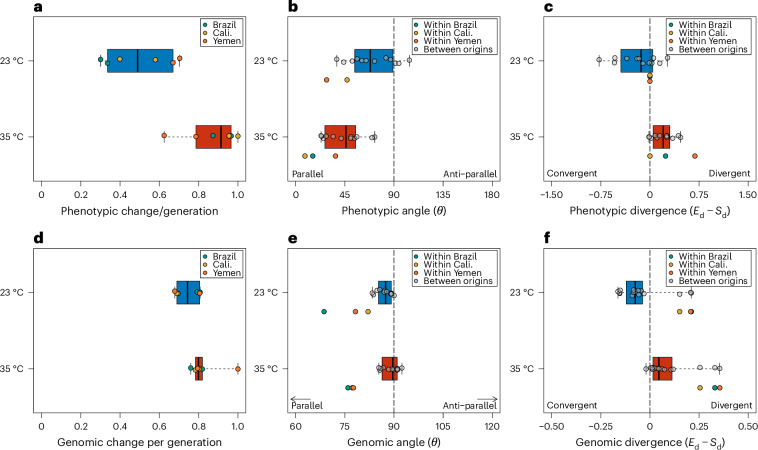
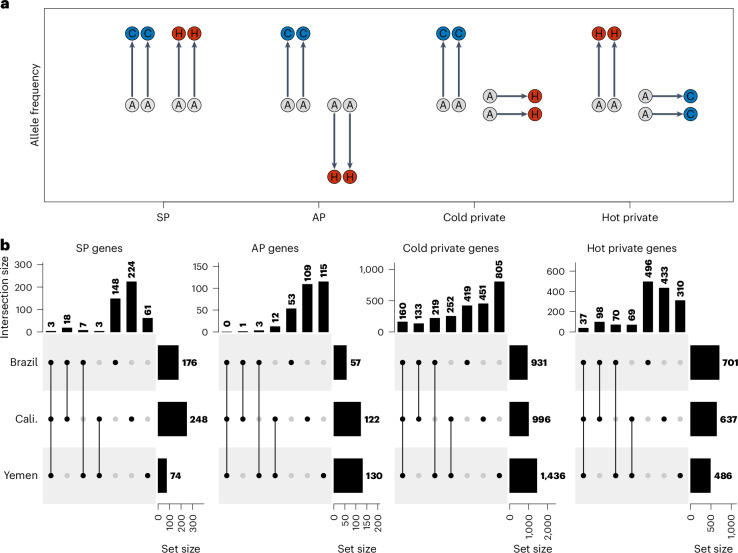
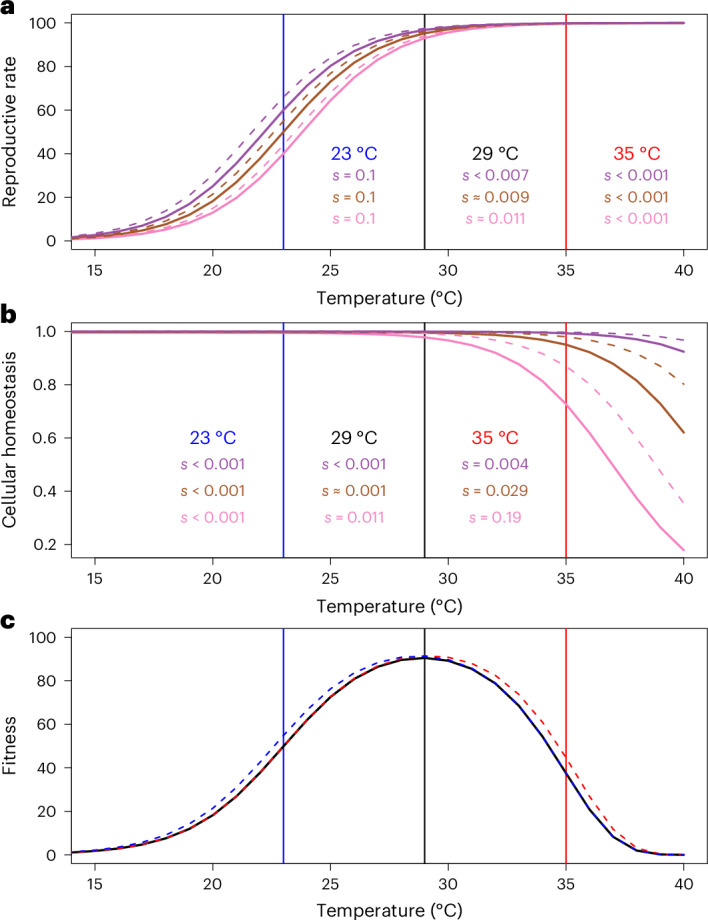
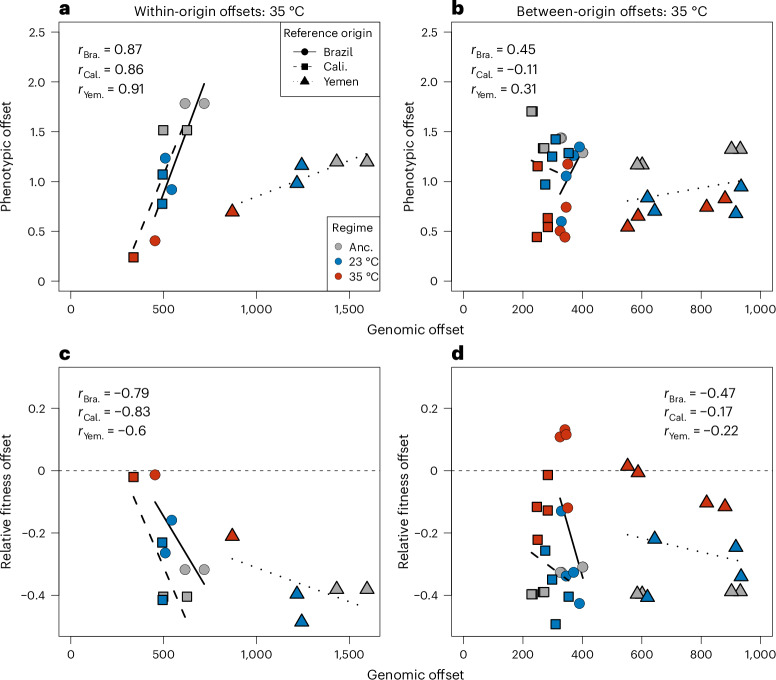
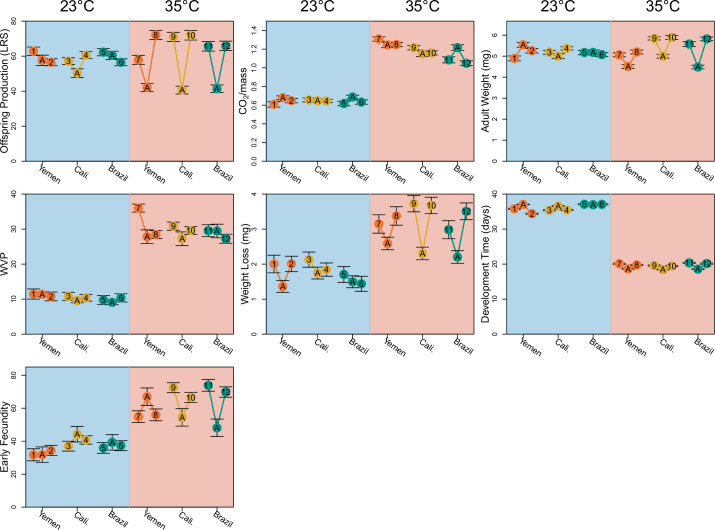
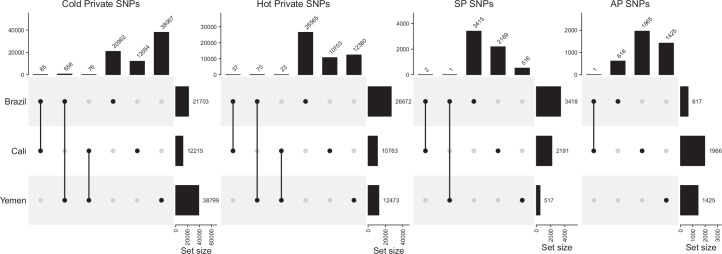
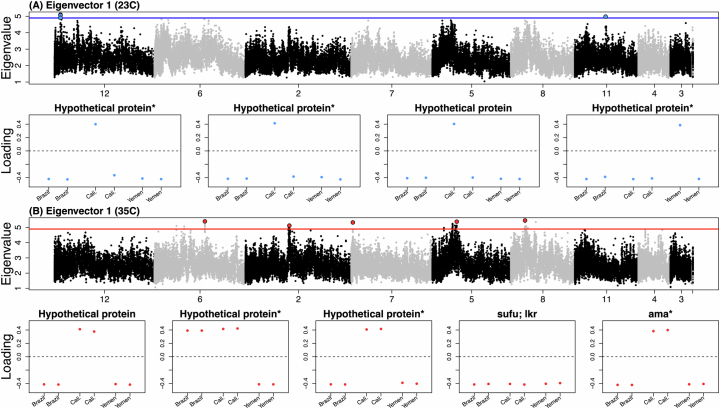
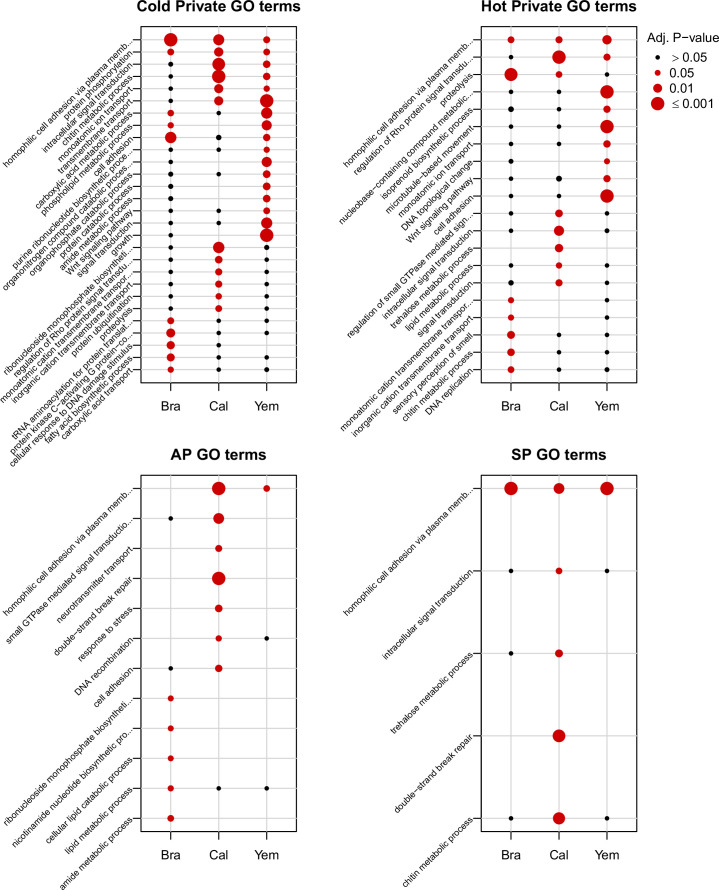
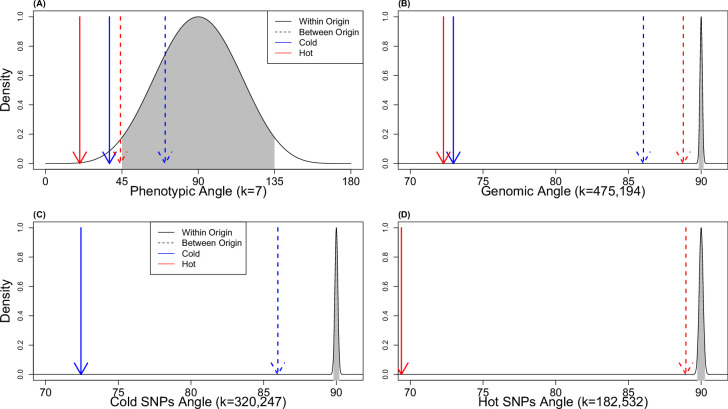
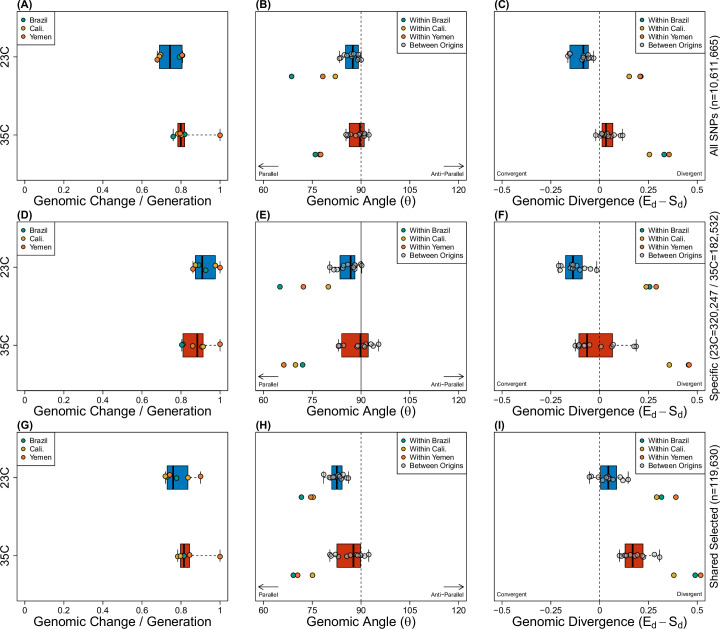
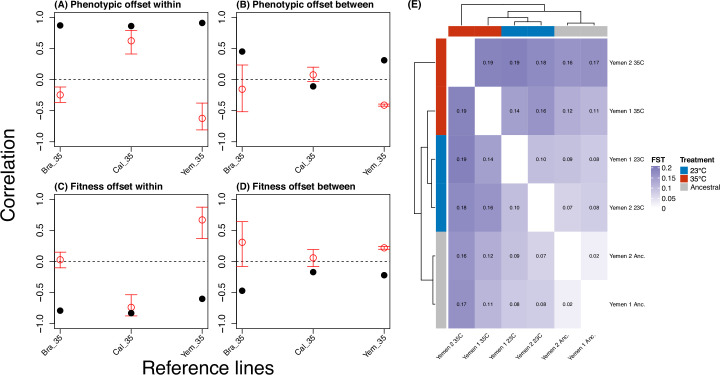
Climate warming is threatening biodiversity by increasing temperatures beyond the optima of many ectotherms. Owing to the inherent non-linear relationship between temperature and the rate of cellular processes, such shifts towards hot temperature are predicted to impose stronger selection compared with corresponding shifts towards cold temperature. This suggests that when adaptation to warming occurs, it should be relatively rapid and predictable. Here we tested this hypothesis from the level of single-nucleotide polymorphisms to life-history traits in the beetle Callosobruchus maculatus. We conducted an evolve-and-resequence experiment on three genetic backgrounds of the beetle reared at hot or cold temperature. Indeed, we find that phenotypic evolution was faster and more repeatable at hot temperature. However, at the genomic level, adaptation to heat was less repeatable when compared across genetic backgrounds. As a result, genomic predictions of phenotypic adaptation in populations exposed to hot temperature were accurate within, but not between, backgrounds. These results seem best explained by genetic redundancy and an increased importance of epistasis during adaptation to heat, and imply that the same mechanisms that exert strong selection and increase repeatability of phenotypic evolution at hot temperature reduce repeatability at the genomic level. Thus, predictions of adaptation in key phenotypes from genomic data may become increasingly difficult as climates warm.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Dynamics of genomic change during evolutionary rescue in the seed beetle Callosobruchus maculatus.Mol Ecol. 2019 May;28(9):2136-2154. doi: 10.1111/mec.15085. Epub 2019 May 2. Mol Ecol. 2019. PMID: 30963641
-
Maladaptive plasticity facilitates evolution of thermal tolerance during an experimental range shift.BMC Evol Biol. 2020 Apr 23;20(1):47. doi: 10.1186/s12862-020-1589-7. BMC Evol Biol. 2020. PMID: 32326878 Free PMC article.
-
Life-history adaptation under climate warming magnifies the agricultural footprint of a cosmopolitan insect pest.Nat Commun. 2025 Jan 18;16(1):827. doi: 10.1038/s41467-025-56177-2. Nat Commun. 2025. PMID: 39827176 Free PMC article.
-
Insect responses to heat: physiological mechanisms, evolution and ecological implications in a warming world.Biol Rev Camb Philos Soc. 2020 Jun;95(3):802-821. doi: 10.1111/brv.12588. Epub 2020 Feb 8. Biol Rev Camb Philos Soc. 2020. PMID: 32035015 Review.
-
Experimental Evolution in a Warming World: The Omics Era.Mol Biol Evol. 2024 Aug 2;41(8):msae148. doi: 10.1093/molbev/msae148. Mol Biol Evol. 2024. PMID: 39034684 Free PMC article. Review.
Cited by
-
Adaptability to climate change is difficult to predict.Nat Ecol Evol. 2025 Jun;9(6):892-893. doi: 10.1038/s41559-025-02731-6. Nat Ecol Evol. 2025. PMID: 40379981 No abstract available.
References
-
- Lässig, M., Mustonen, V. & Walczak, A. M. Predicting evolution. Nat. Ecol. Evol.1, 0077 (2017). - PubMed
-
- Blount, Z. D., Lenski, R. E. & Losos, J. B. Contingency and determinism in evolution: replaying life’s tape. Science362, eaam5979 (2018). - PubMed
-
- Svensson, E. I. & Berger, D. The role of mutation bias in adaptive evolution. Trends Ecol. Evol.34, 422–434 (2019). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
