

Dynamic changes in macrophage populations and resulting alterations in Prostaglandin E2 sensitivity in mice with diet-induced MASH
- PMID: 40380177
- PMCID: PMC12083000
- DOI: 10.1186/s12964-025-02222-y
Dynamic changes in macrophage populations and resulting alterations in Prostaglandin E2 sensitivity in mice with diet-induced MASH
Abstract
Background: The transition from metabolic dysfunction-associated steatotic liver disease (MASLD) to steatohepatitis (MASH) is characterized by a chronic low-grade inflammation, involving activation of resident macrophages (Kupffer cells; KC) and recruitment of infiltrating macrophages. Macrophages produce cytokines and, after induction of Cyclooxygenase 2 (COX-2), the key enzyme of prostanoid synthesis, prostaglandin E2 (PGE2). PGE2 modulates cytokine production in an autocrine and paracrine manner, therefore playing a pivotal role in regulating inflammatory processes. Changes in the hepatic macrophage pool during MASLD progression to MASH could influence PGE2- and cytokine-mediated signaling processes. The aim of this study was to characterize these changes in mice with diet-induced MASH and further elucidate the role of COX-2-dependently formed PGE2 on the inflammatory response in different macrophage populations of mice with a macrophage-specific COX-2-deletion.
Methods: Male, 6-7-week-old wildtype mice were fed either a Standard or high-fat, high-cholesterol MASH-inducing diet for 4, 12 and 20 weeks. Liver macrophages were isolated and analyzed by flow cytometry. For in vitro experiments primary KC, peritoneal macrophages (PM) and Bone-marrow-derived macrophages (BMDM) were isolated from macrophage-specific COX-2-deficient and wildtype mice and treated with lipopolysaccharide (LPS) and/or PGE2.
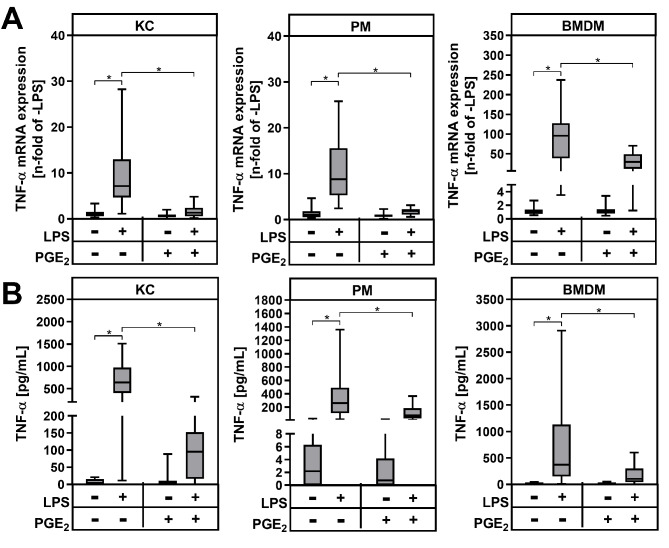
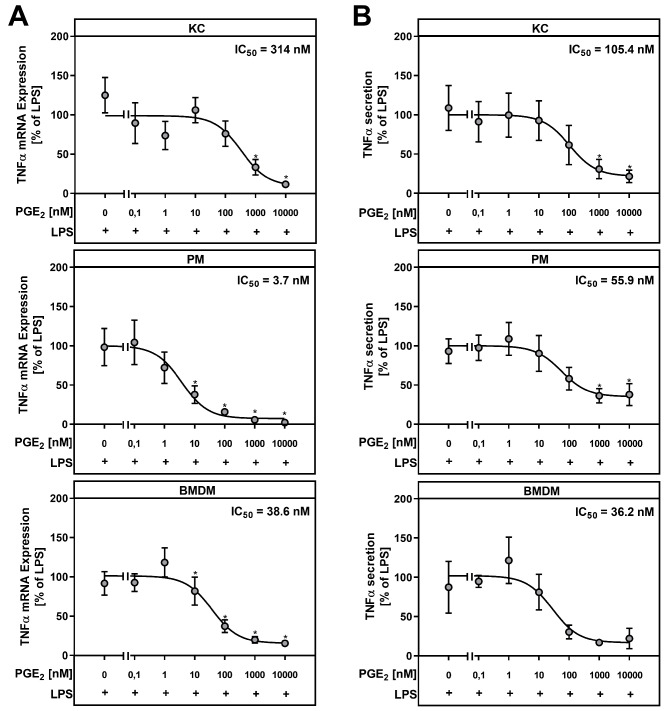
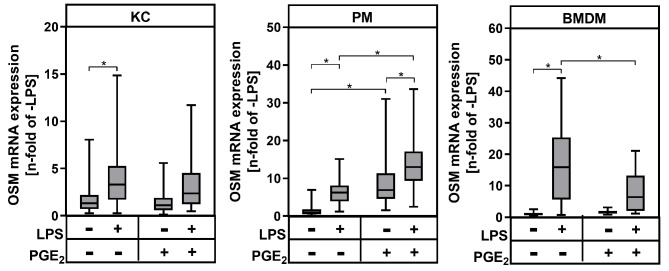
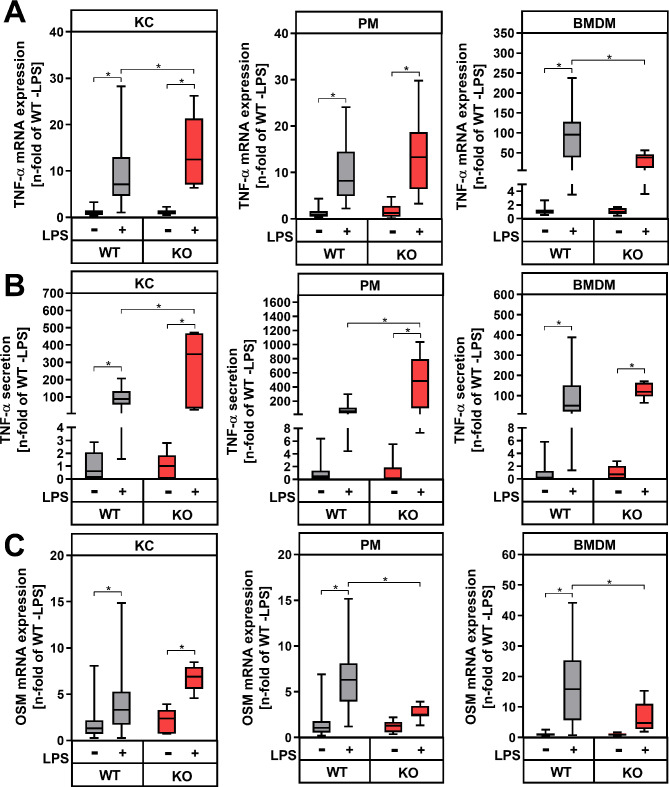
Results: During MASH-development, the proportion of KC (Clec4F+Tim4+) decreased, while the proportion of monocyte-derived macrophages (Clec4F-Tim4-) and monocyte-derived cells exhibiting a phenotype similar to KC (Clec4F+Tim4-) significantly increased over time. In vitro experiments showed that exogenous PGE2 completely abrogated the LPS-induced mRNA expression and secretion of tumor necrosis factor-alpha (TNF-α) in primary KC, PM and BMDM from wildtype mice. PM and BMDM, as in vitro models for infiltrating macrophages, were more sensitive to PGE2 compared to KC. Deletion of COX-2 in all macrophage populations led to an impaired PGE2-dependent feedback inhibition of TNF-α production. LPSinduced TNF-α mRNA expression was higher compared to the respective wildtype macrophage population.
Conclusion: The current study, using a murine MASH model, indicates that PGE2 may have a protective, anti-inflammatory effect, especially by inhibiting the expression of pro-inflammatory cytokines such as TNFα in infiltrating monocyte-derived macrophages. An inhibition of endogenous PGE2 synthesis in macrophages by pharmacological inhibition of COX-2 could potentially increase inflammation and promote the progression of MASH.
Keywords: Cyclooxygenase; Infiltrating macrophages; Inflammation; Kupffer cells; MASH; Prostaglandin E2; TNF-α.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures


References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
