

An integrated proteomics approach identifies phosphorylation sites on viral and host proteins that regulate West Nile virus infection
- PMID: 40381193
- PMCID: PMC12180539
- DOI: 10.1016/j.celrep.2025.115728
An integrated proteomics approach identifies phosphorylation sites on viral and host proteins that regulate West Nile virus infection
Abstract
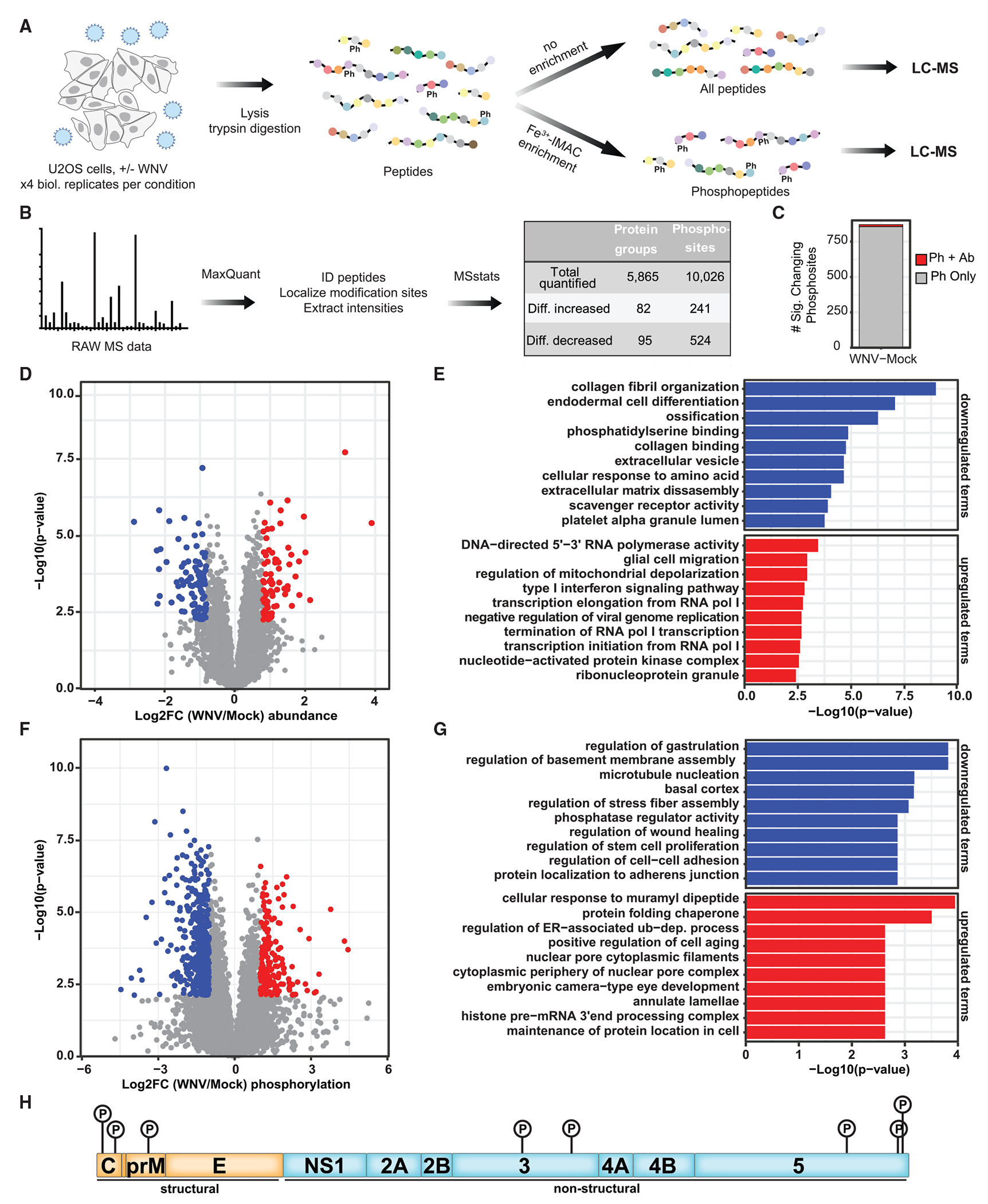
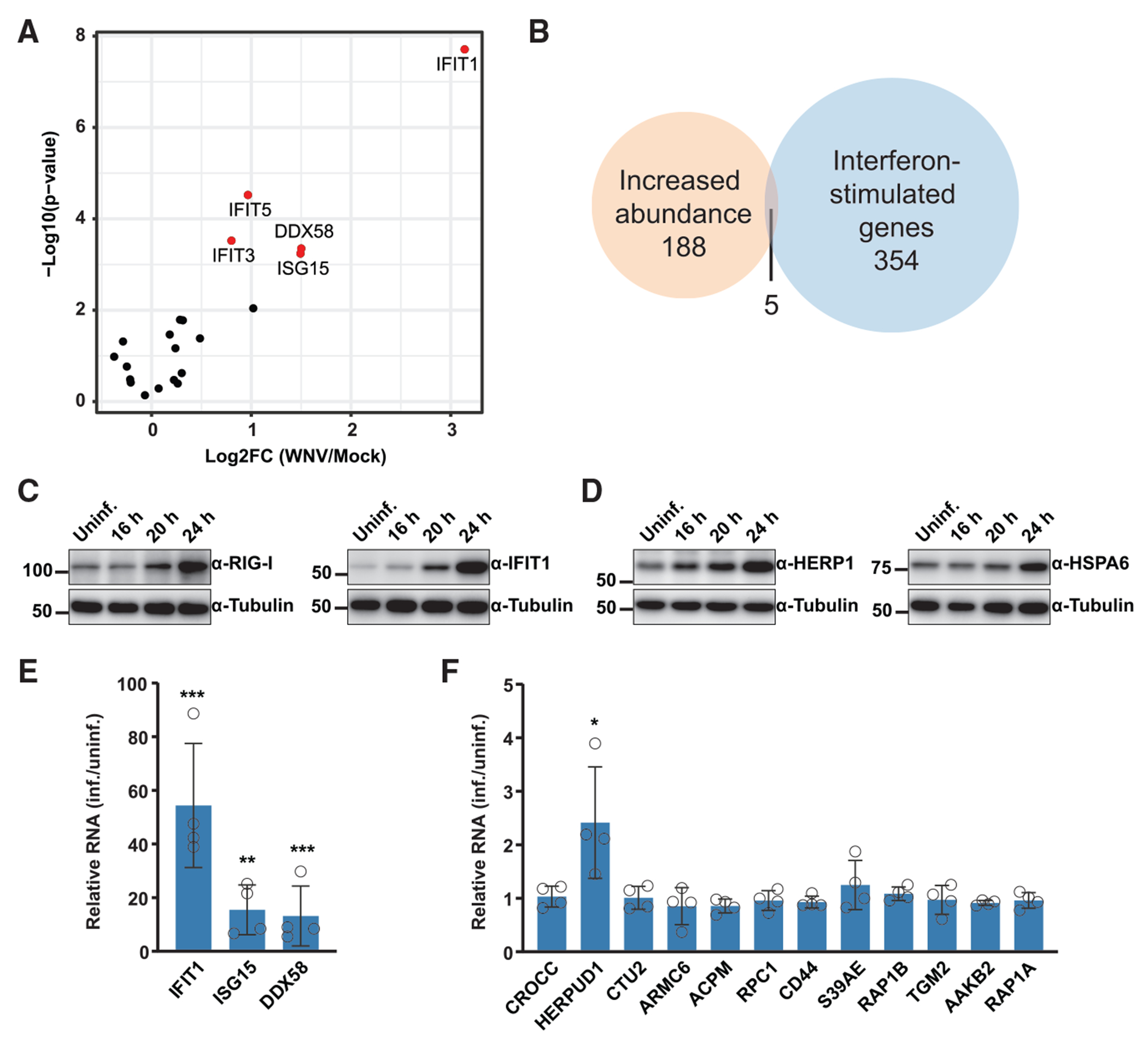
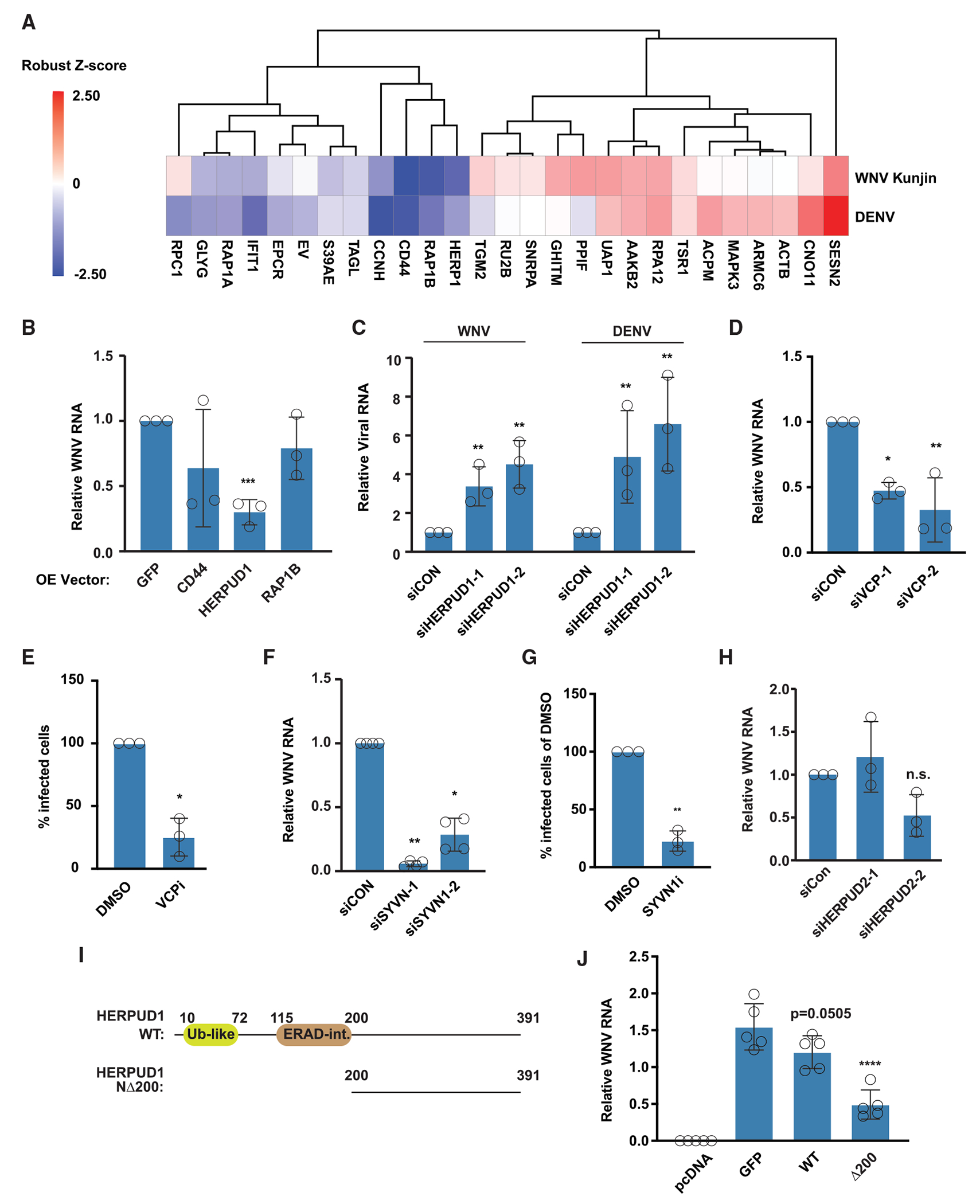
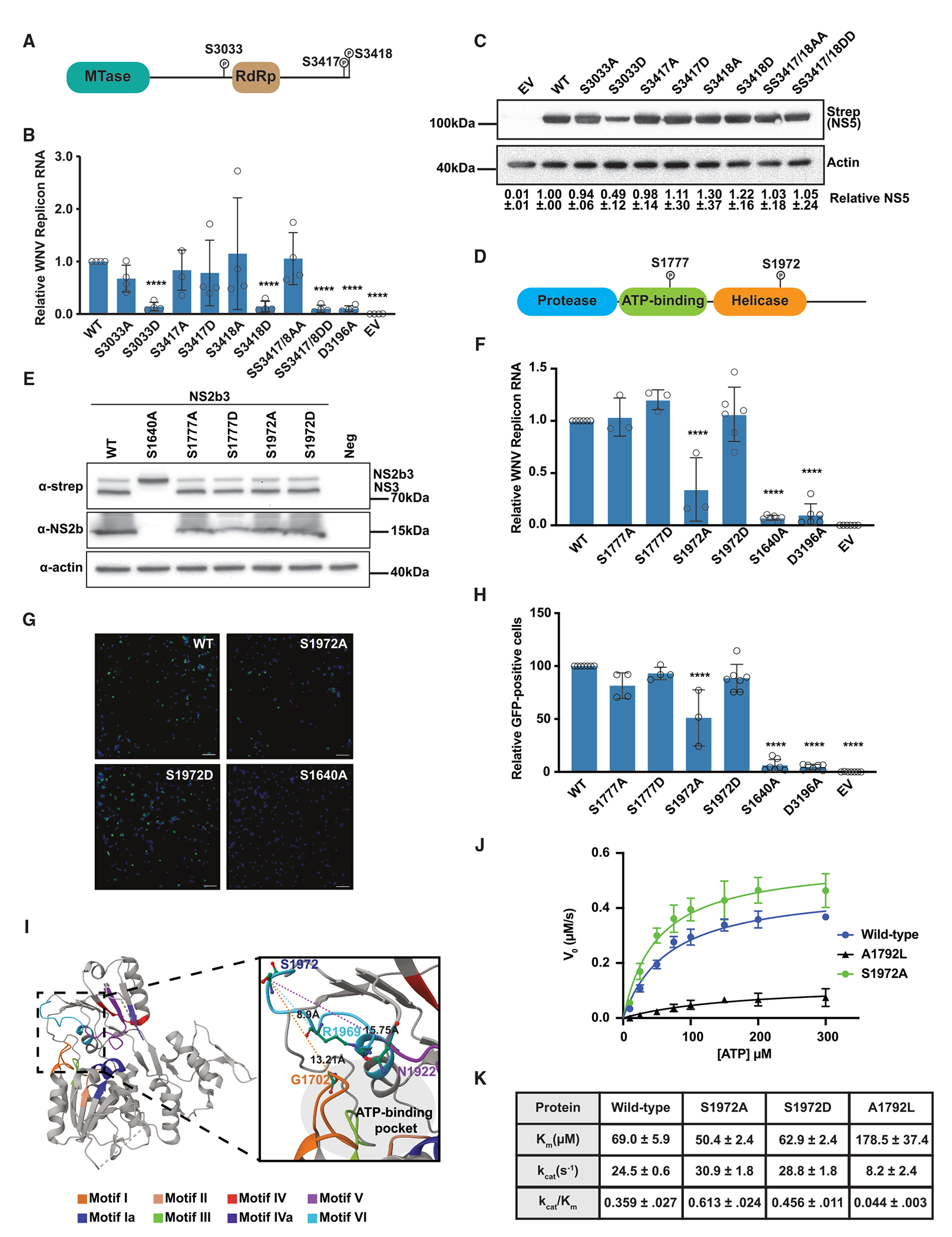
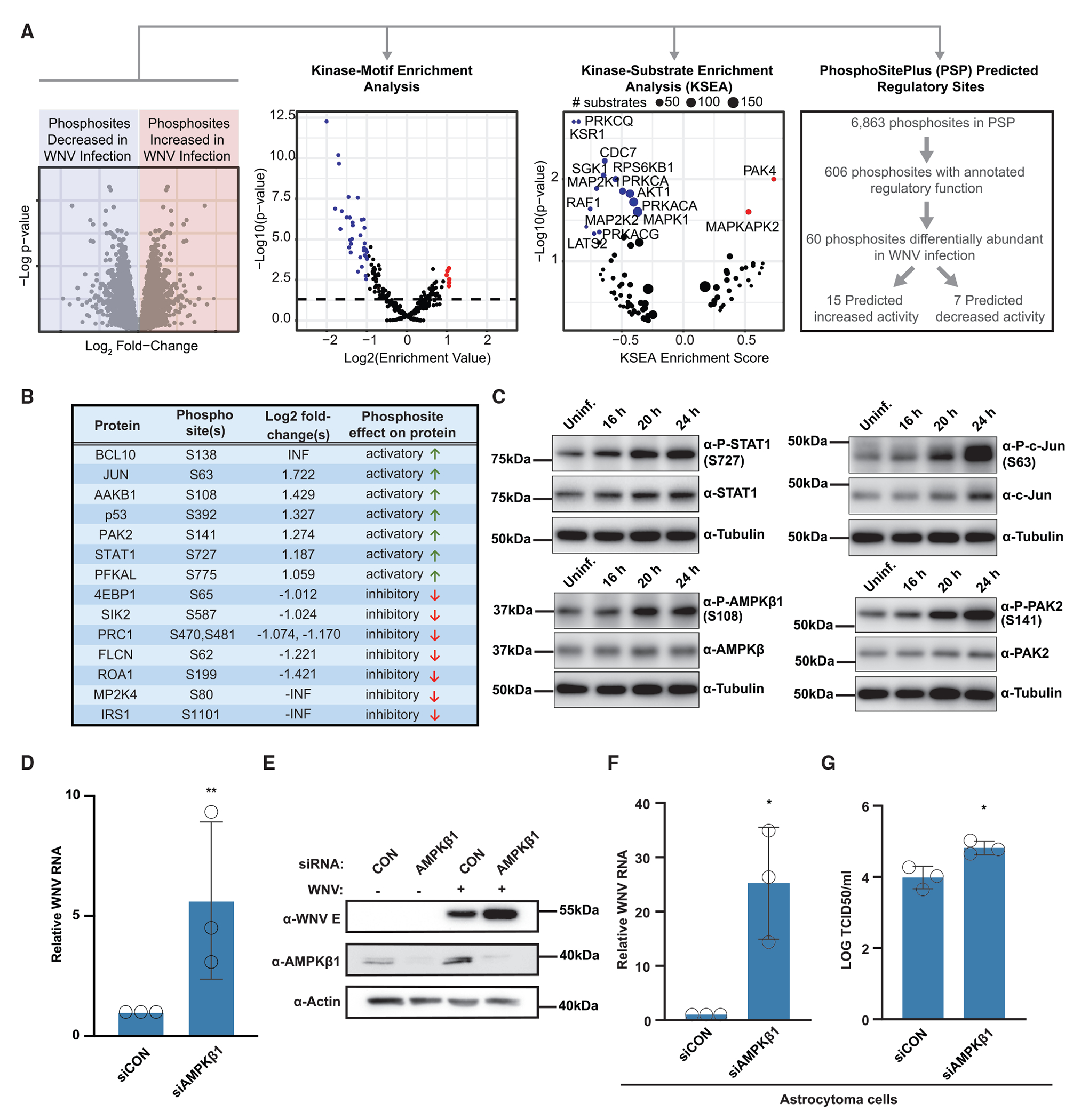
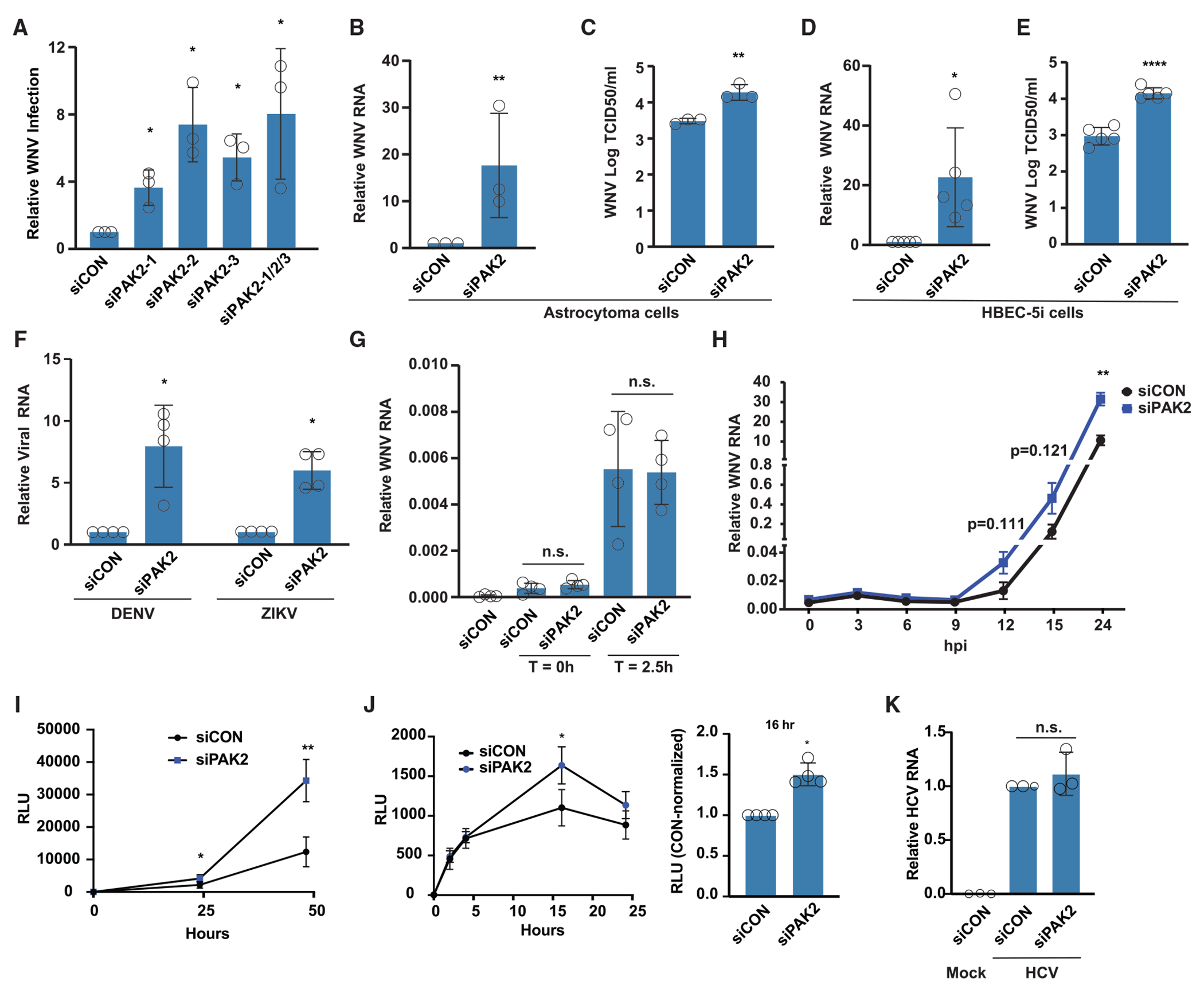
Upon infection, viruses alter the proteome, creating a hospitable environment for infection. Cells respond to limit viral replication, including through protein regulation by post-translational modifications. We use mass spectrometry to define proteome alterations during West Nile virus (WNV) infection. Our studies identify upregulation of HERPUD1, which restricts WNV replication through a mechanism independent of its role in endoplasmic reticulum (ER)-associated degradation (ERAD). We also identify modifications on viral proteins, including a WNV NS3 phosphorylation site that impacts viral replication. Finally, we reveal activation of two host kinases with antiviral activity. We identify phosphorylation at S108 of AMPKβ1, a non-catalytic subunit that regulates activity of the AMPK complex. We also show activation of PAK2 by phosphorylation at S141, which restricts translation of the viral genome. This work contributes to our understanding of the interplay between host and virus while providing a resource to define the changes to the proteome that regulate viral infection.
Keywords: CP: Microbiology; Orthoflaviviruses; West Nile virus; innate immunity; phosphorylation; post-translational modifications; viral helicase; viral translation; virus-host interactions.
Copyright © 2025 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Similar articles
-
UFMylation promotes orthoflavivirus infectious particle production.J Virol. 2025 Jul 22;99(7):e0065425. doi: 10.1128/jvi.00654-25. Epub 2025 Jun 3. J Virol. 2025. PMID: 40459261 Free PMC article.
-
Towards a Universal Translator: Decoding the PTMs That Regulate Orthoflavivirus Infection.Viruses. 2025 Feb 19;17(2):287. doi: 10.3390/v17020287. Viruses. 2025. PMID: 40007042 Free PMC article. Review.
-
Adenovirus Modulates Toll-Like Receptor 4 Signaling by Reprogramming ORP1L-VAP Protein Contacts for Cholesterol Transport from Endosomes to the Endoplasmic Reticulum.J Virol. 2017 Feb 28;91(6):e01904-16. doi: 10.1128/JVI.01904-16. Print 2017 Mar 15. J Virol. 2017. PMID: 28077646 Free PMC article.
-
Classical swine fever virus utilizes stearoyl-CoA desaturase 1-mediated lipid metabolism to facilitate viral replication.J Virol. 2025 Jun 17;99(6):e0055125. doi: 10.1128/jvi.00551-25. Epub 2025 May 19. J Virol. 2025. PMID: 40387378 Free PMC article.
-
Structured treatment interruptions (STI) in chronic unsuppressed HIV infection in adults.Cochrane Database Syst Rev. 2006 Jul 19;2006(3):CD006148. doi: 10.1002/14651858.CD006148. Cochrane Database Syst Rev. 2006. PMID: 16856117 Free PMC article.
Cited by
-
Yellow Fever Virus Interactomes Reveal Common and Divergent Strategies of Replication and Evolution for Mosquito-borne Flaviviruses.bioRxiv [Preprint]. 2025 Jul 1:2025.06.14.659623. doi: 10.1101/2025.06.14.659623. bioRxiv. 2025. PMID: 40631121 Free PMC article. Preprint.
-
Allosteric Regulation of RNA Affinity by Motif V-VI Coupling in West Nile Virus NS3 Helicase.bioRxiv [Preprint]. 2025 Jul 4:2025.07.04.663198. doi: 10.1101/2025.07.04.663198. bioRxiv. 2025. PMID: 40631148 Free PMC article. Preprint.
References
-
- Li M, Johnson JR, Truong B, Kim G, Weinbren N, Dittmar M, Shah PS, Von Dollen J, Newton BW, Jang GM, et al. (2019). Identification of antiviral roles for the exon-junction complex and nonsense-mediated decay in flaviviral infection. Nat. Microbiol 4, 985–995. 10.1038/s41564-019-0375-z. - DOI - PMC - PubMed
-
- Besson B, Basset J, Gatellier S, Chabrolles H, Chaze T, Hourdel V, Matondo M, Pardigon N, and Choumet V (2020). Comparison of a human neuronal model proteome upon Japanese encephalitis or West Nile Virus infection and potential role of mosquito saliva in neuropathogenesis. PLoS One 15, e0232585. 10.1371/journal.pone.0232585. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
