

CREATE: cell-type-specific cis-regulatory element identification via discrete embedding
- PMID: 40382355
- PMCID: PMC12085597
- DOI: 10.1038/s41467-025-59780-5
CREATE: cell-type-specific cis-regulatory element identification via discrete embedding
Abstract
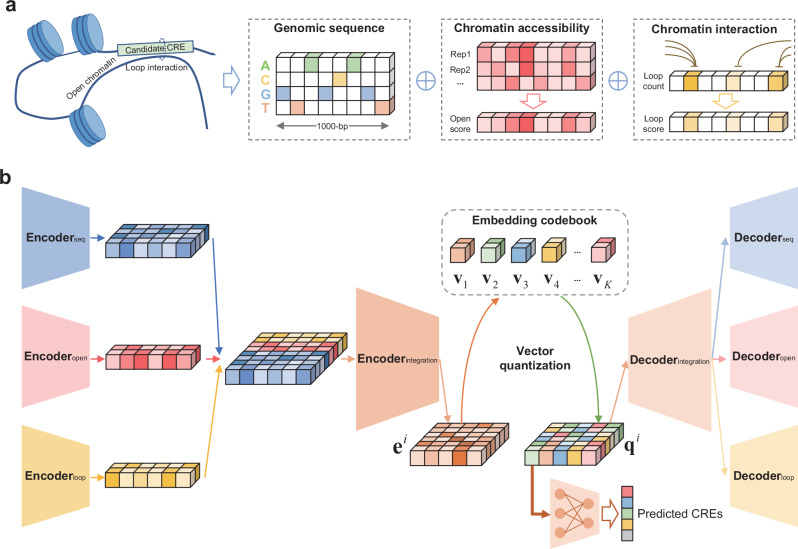
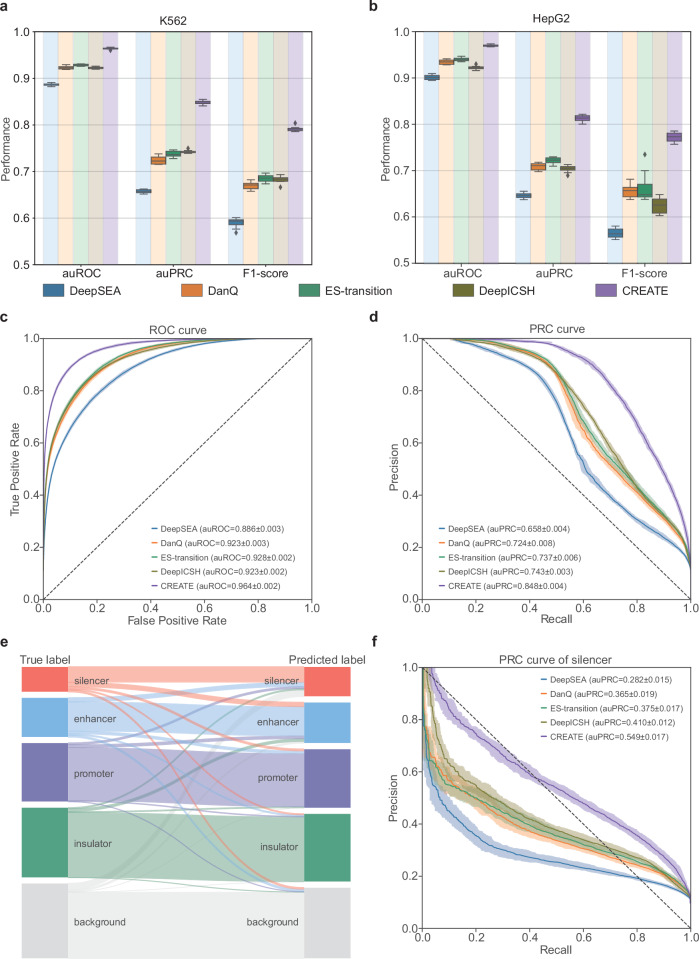
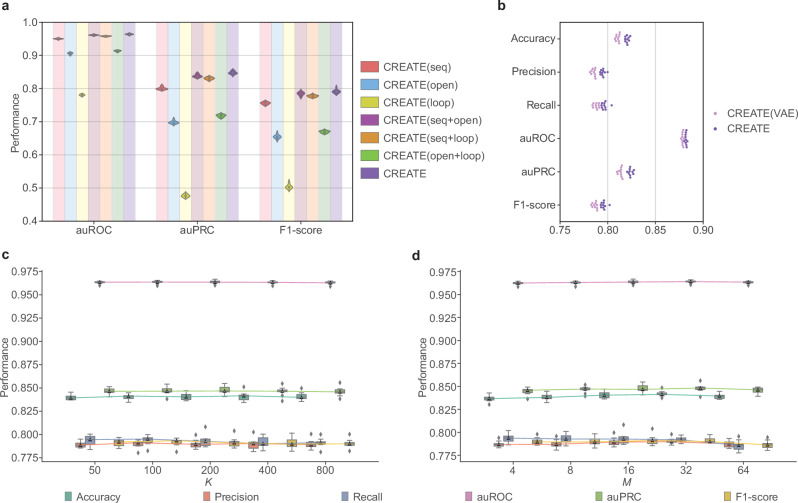
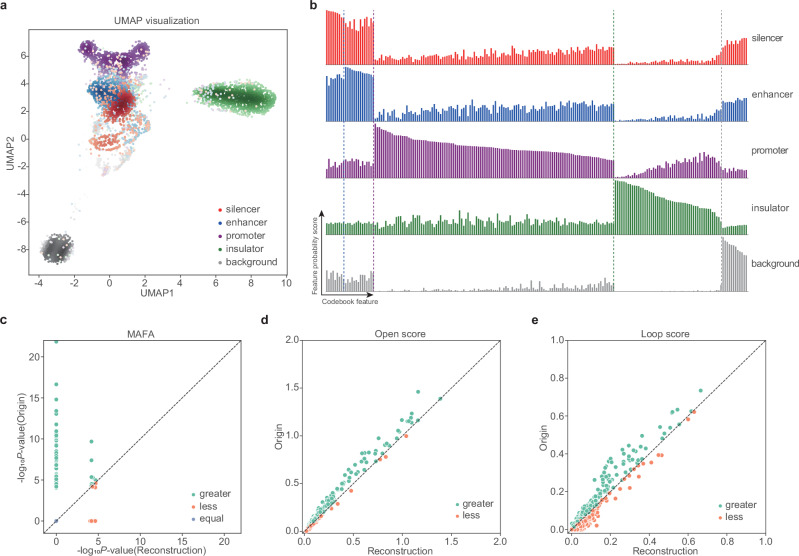
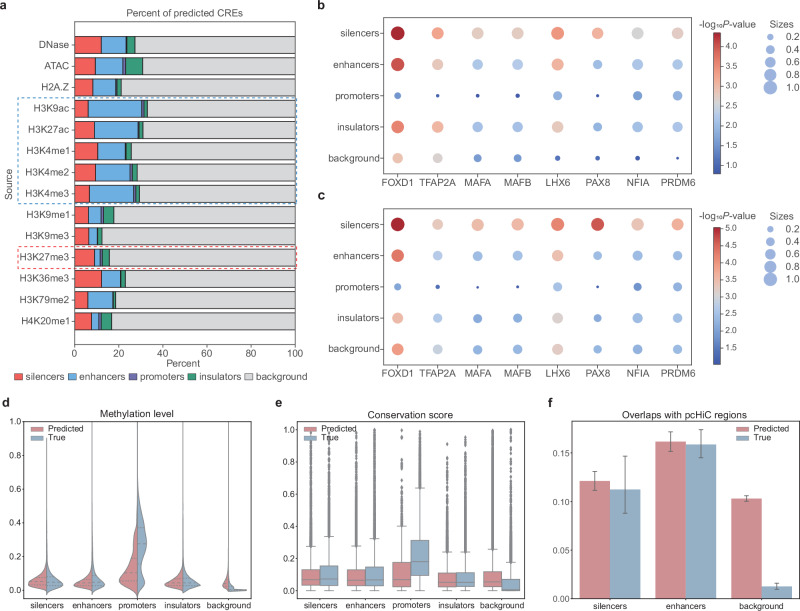
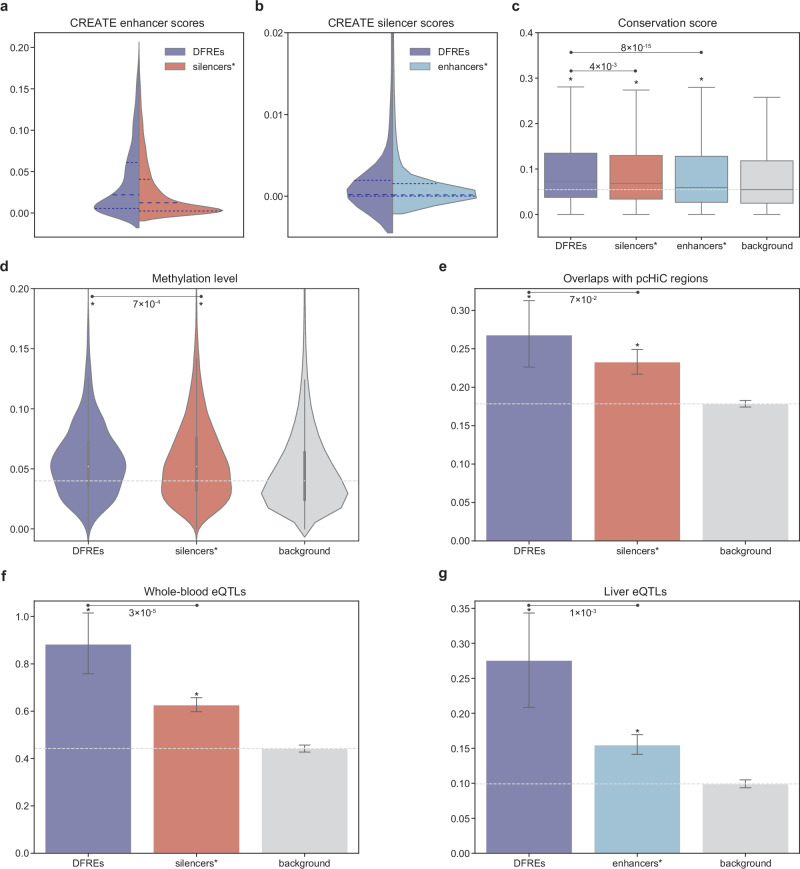
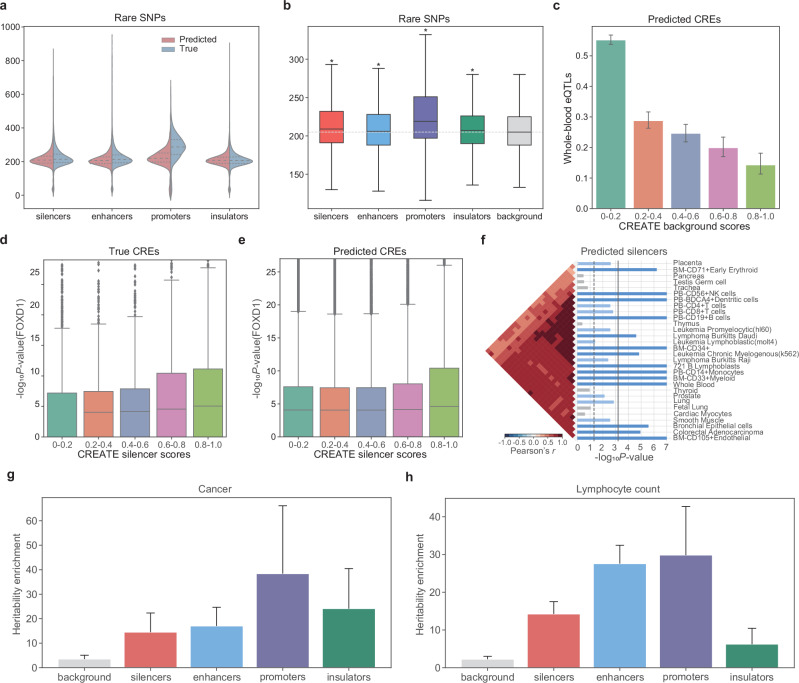
Cis-regulatory elements (CREs), including enhancers, silencers, promoters and insulators, play pivotal roles in orchestrating gene regulatory mechanisms that drive complex biological traits. However, current approaches for CRE identification are predominantly sequence-based and typically focus on individual CRE types, limiting insights into their cell-type-specific functions and regulatory dynamics. Here, we present CREATE, a multimodal deep learning framework based on Vector Quantized Variational AutoEncoder, tailored for comprehensive CRE identification and characterization. CREATE integrates genomic sequences, chromatin accessibility, and chromatin interaction data to generate discrete CRE embeddings, enabling accurate multi-class classification and robust characterization of CREs. CREATE excels in identifying cell-type-specific CREs, and provides quantitative and interpretable insights into CRE-specific features, uncovering the underlying regulatory codes. By facilitating large-scale prediction of CREs in specific cell types, CREATE enhances the recognition of disease- or phenotype-associated biological variabilities of CREs, thus advancing our understanding of gene regulatory landscapes and their roles in health and disease.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
A novel pairwise comparison method for in silico discovery of statistically significant cis-regulatory elements in eukaryotic promoter regions: application to Arabidopsis.J Theor Biol. 2015 Jan 7;364:364-76. doi: 10.1016/j.jtbi.2014.09.038. Epub 2014 Oct 7. J Theor Biol. 2015. PMID: 25303887
-
Integrative modeling of eQTLs and cis-regulatory elements suggests mechanisms underlying cell type specificity of eQTLs.PLoS Genet. 2013;9(8):e1003649. doi: 10.1371/journal.pgen.1003649. Epub 2013 Aug 1. PLoS Genet. 2013. PMID: 23935528 Free PMC article.
-
DNA regulatory element cooperation and competition in transcription.BMB Rep. 2024 Dec;57(12):509-520. doi: 10.5483/BMBRep.2024-0069. BMB Rep. 2024. PMID: 39523506 Free PMC article. Review.
-
Multiscale footprints reveal the organization of cis-regulatory elements.Nature. 2025 Feb;638(8051):779-786. doi: 10.1038/s41586-024-08443-4. Epub 2025 Jan 22. Nature. 2025. PMID: 39843737 Free PMC article.
-
Identification, Design, and Application of Noncoding Cis-Regulatory Elements.Biomolecules. 2024 Aug 5;14(8):945. doi: 10.3390/biom14080945. Biomolecules. 2024. PMID: 39199333 Free PMC article. Review.
References
-
- Berger, S. L. The complex language of chromatin regulation during transcription. Nature447, 407–412 (2007). - PubMed
-
- Maston, G. A., Evans, S. K. & Green, M. R. Transcriptional regulatory elements in the human genome. Annu. Rev. Genomics Hum. Genet.7, 29–59 (2006). - PubMed
-
- Chatterjee, S. & Ahituv, N. Gene regulatory elements, major drivers of human disease. Annu. Rev. Genomics Hum. Genet.18, 45–63 (2017). - PubMed
-
- Klemm, S. L., Shipony, Z. & Greenleaf, W. J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet.20, 207–220 (2019). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
