

Immune perturbations in human pancreas lymphatic tissues prior to and after type 1 diabetes onset
- PMID: 40383826
- PMCID: PMC12086209
- DOI: 10.1038/s41467-025-59626-0
Immune perturbations in human pancreas lymphatic tissues prior to and after type 1 diabetes onset
Abstract
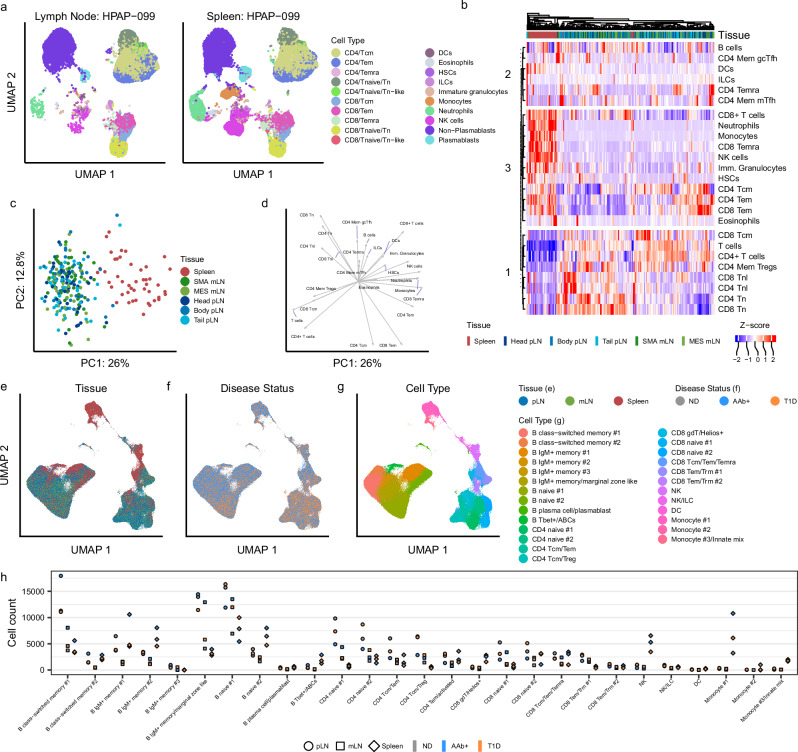
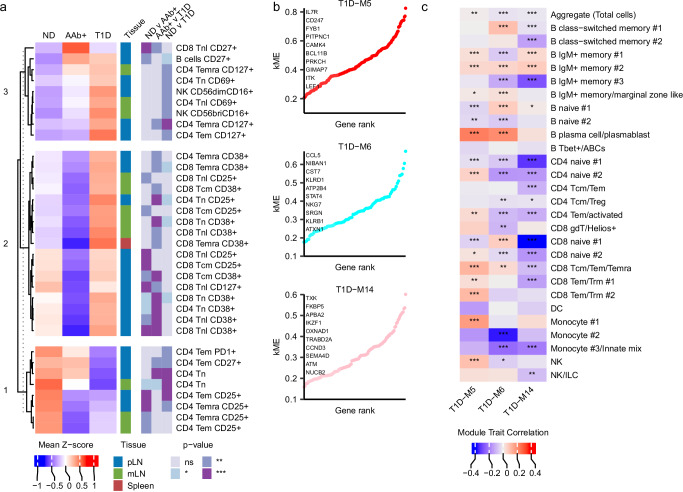
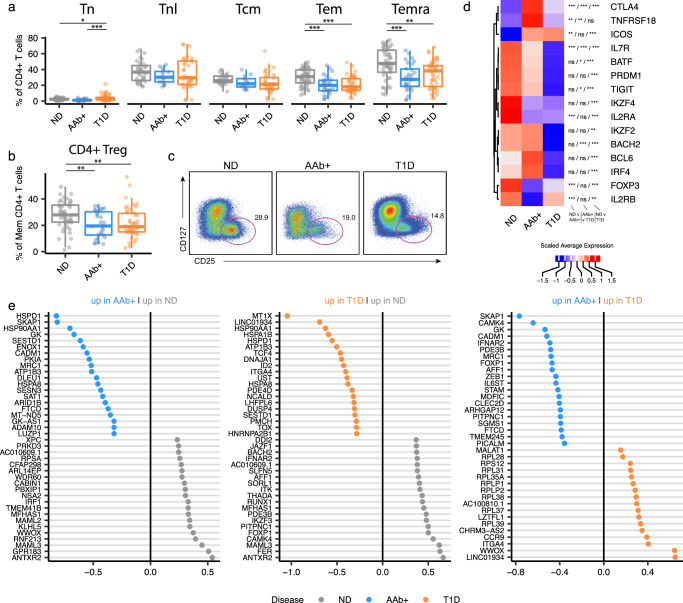
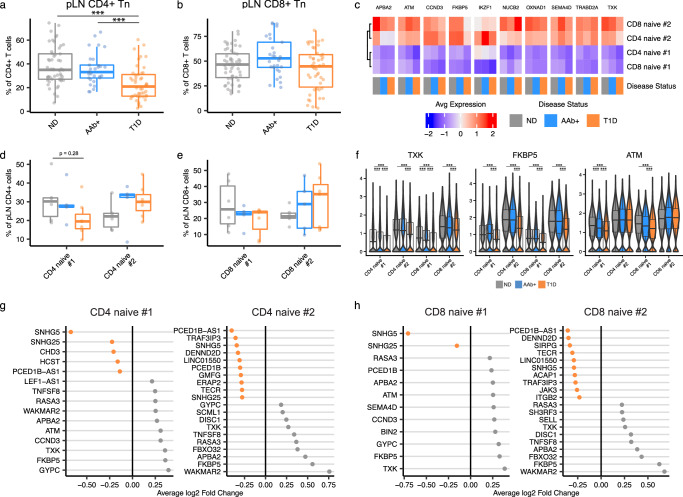
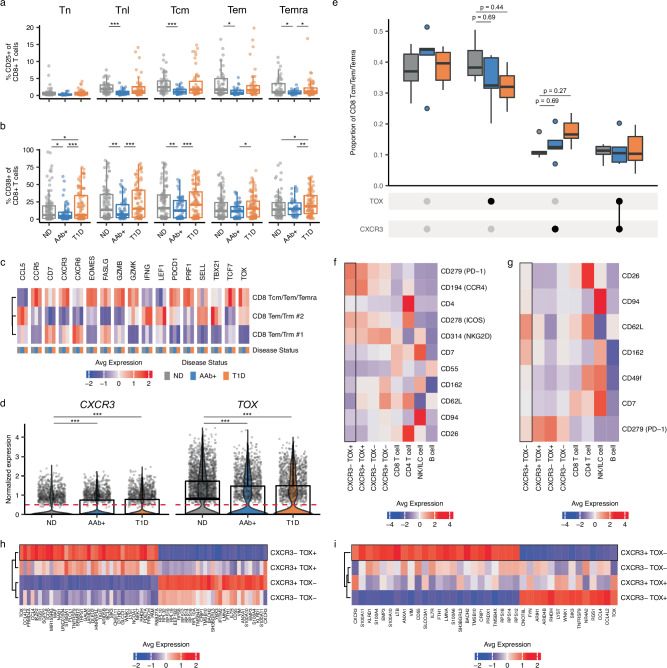
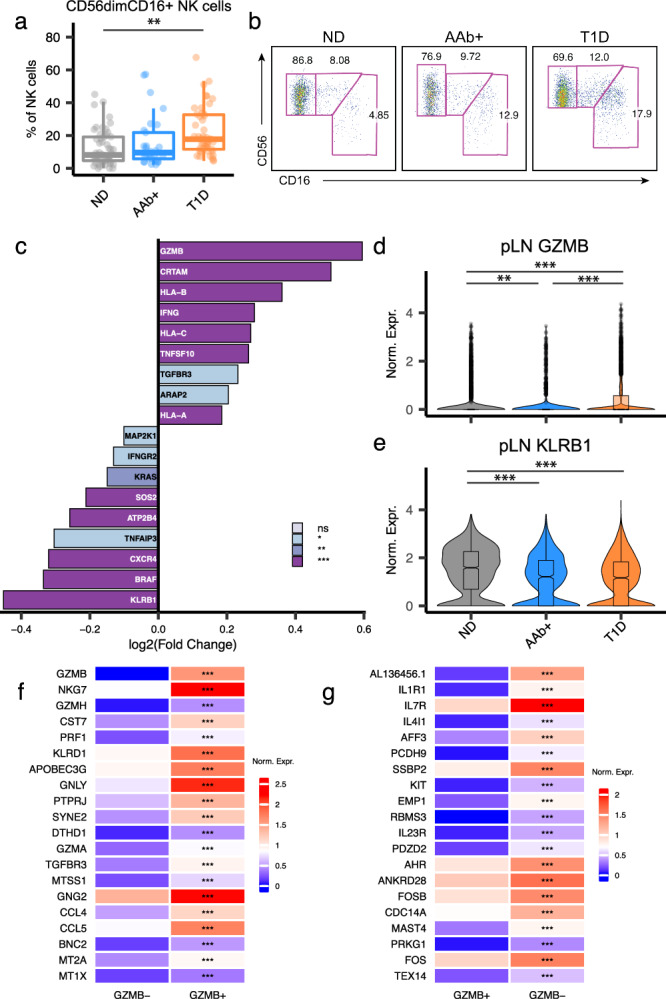
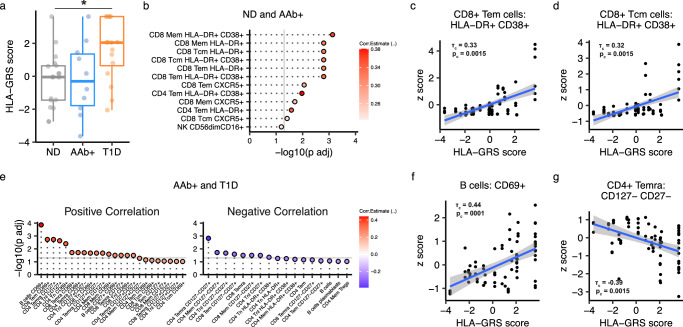
Autoimmune destruction of pancreatic β cells results in type 1 diabetes (T1D), with pancreatic immune infiltrate representing a key feature in this process. However, characterization of the immunological processes occurring in human pancreatic lymphatic tissues is lacking. Here, we conduct a comprehensive study of immune cells from pancreatic, mesenteric, and splenic lymphatic tissues of non-diabetic control (ND), β cell autoantibody-positive non-diabetic (AAb+), and T1D donors using flow cytometry and CITEseq. Compared to ND pancreas-draining lymph nodes (pLN), AAb+ and T1D donor pLNs display decreased CD4+ Treg and increased stem-like CD8+ T cell signatures, while only T1D donor pLNs exhibit naive T cell and NK cell differentiation. Mesenteric LNs have modulations only in CD4+ Tregs and naive cells, while splenocytes lack these perturbations. Further, T cell expression of activation markers and IL7 receptor correlate with T1D genetic risk. These results demonstrate tissue-restricted immune changes occur before and after T1D onset.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Update of
-
Immune perturbations in human pancreas lymphatic tissues prior to and after type 1 diabetes onset.bioRxiv [Preprint]. 2024 Sep 16:2024.04.23.590798. doi: 10.1101/2024.04.23.590798. bioRxiv. 2024. Update in: Nat Commun. 2025 May 18;16(1):4621. doi: 10.1038/s41467-025-59626-0. PMID: 39345402 Free PMC article. Updated. Preprint.
Similar articles
-
The extra-islet pancreas supports autoimmunity in human type 1 diabetes.Elife. 2025 Apr 15;13:RP100535. doi: 10.7554/eLife.100535. Elife. 2025. PMID: 40232951 Free PMC article.
-
Preproinsulin-specific regulatory T cell-derived exosomes loaded with immune-checkpoint ligands can suppress autoimmune responses in type 1 diabetes.Int Immunopharmacol. 2025 Aug 28;161:115045. doi: 10.1016/j.intimp.2025.115045. Epub 2025 Jun 7. Int Immunopharmacol. 2025. PMID: 40482450
-
Immune perturbations in human pancreas lymphatic tissues prior to and after type 1 diabetes onset.bioRxiv [Preprint]. 2024 Sep 16:2024.04.23.590798. doi: 10.1101/2024.04.23.590798. bioRxiv. 2024. Update in: Nat Commun. 2025 May 18;16(1):4621. doi: 10.1038/s41467-025-59626-0. PMID: 39345402 Free PMC article. Updated. Preprint.
-
The emerging relationship between mucosal-associated invariant T cell populations and the onset and progression of type 1 diabetes.Front Immunol. 2025 Jun 18;16:1602934. doi: 10.3389/fimmu.2025.1602934. eCollection 2025. Front Immunol. 2025. PMID: 40607421 Free PMC article. Review.
-
TNF-α inhibitors for type 1 diabetes: exploring the path to a pivotal clinical trial.Front Immunol. 2024 Oct 1;15:1470677. doi: 10.3389/fimmu.2024.1470677. eCollection 2024. Front Immunol. 2024. PMID: 39411715 Free PMC article. Review.
Cited by
-
Pharmacological inhibition of tyrosine protein-kinase 2 reduces islet inflammation and delays type 1 diabetes onset in mice.EBioMedicine. 2025 Jul;117:105734. doi: 10.1016/j.ebiom.2025.105734. Epub 2025 May 6. EBioMedicine. 2025. PMID: 40335415 Free PMC article.
-
Profiling associations of interactive ligand-receptors (HLA class I and KIR gene products) with the progression to type 1 diabetes among seroconverted participants.Diabetologia. 2025 Aug 21. doi: 10.1007/s00125-025-06520-5. Online ahead of print. Diabetologia. 2025. PMID: 40835749
-
Spatial transcriptomics from pancreas and local draining lymph node tissue reveals a lymphotoxin-β signature in human type 1 diabetes.bioRxiv [Preprint]. 2025 May 30:2025.05.19.654940. doi: 10.1101/2025.05.19.654940. bioRxiv. 2025. PMID: 40475580 Free PMC article. Preprint.
References
MeSH terms
Substances
Grants and funding
- P01 AI042288/AI/NIAID NIH HHS/United States
- UC4-DK-112217/U.S. Department of Health & Human Services | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (National Institute of Diabetes & Digestive & Kidney Diseases)
- UC4 DK112232/DK/NIDDK NIH HHS/United States
- U01 DK123594/DK/NIDDK NIH HHS/United States
- 5-P30-DK-019525/U.S. Department of Health & Human Services | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (National Institute of Diabetes & Digestive & Kidney Diseases)
LinkOut - more resources
Full Text Sources
Medical
Research Materials
