

A quantitative comparison of the deleteriousness of missense and nonsense mutations using the structurally resolved human protein interactome
- PMID: 40384578
- PMCID: PMC12086521
- DOI: 10.1002/pro.70155
A quantitative comparison of the deleteriousness of missense and nonsense mutations using the structurally resolved human protein interactome
Abstract
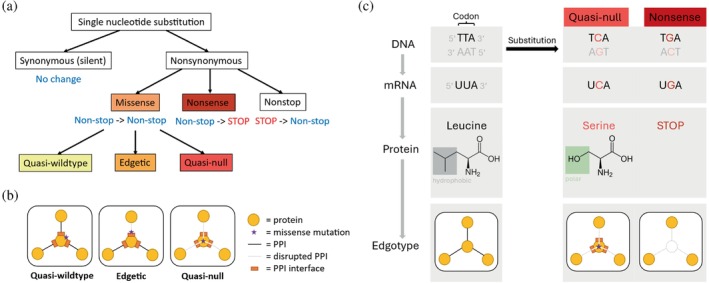
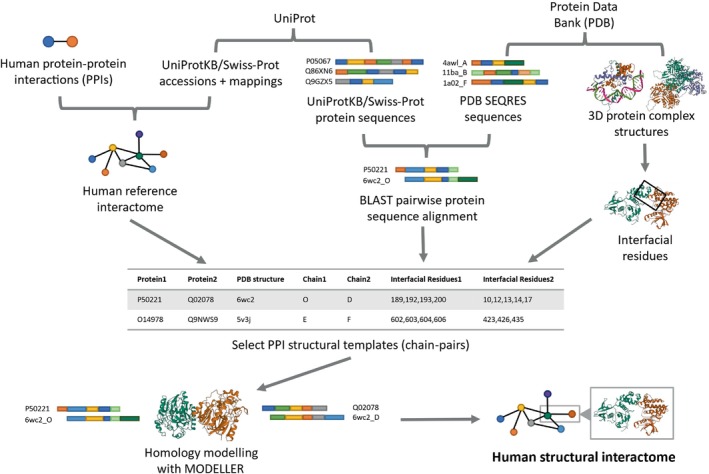
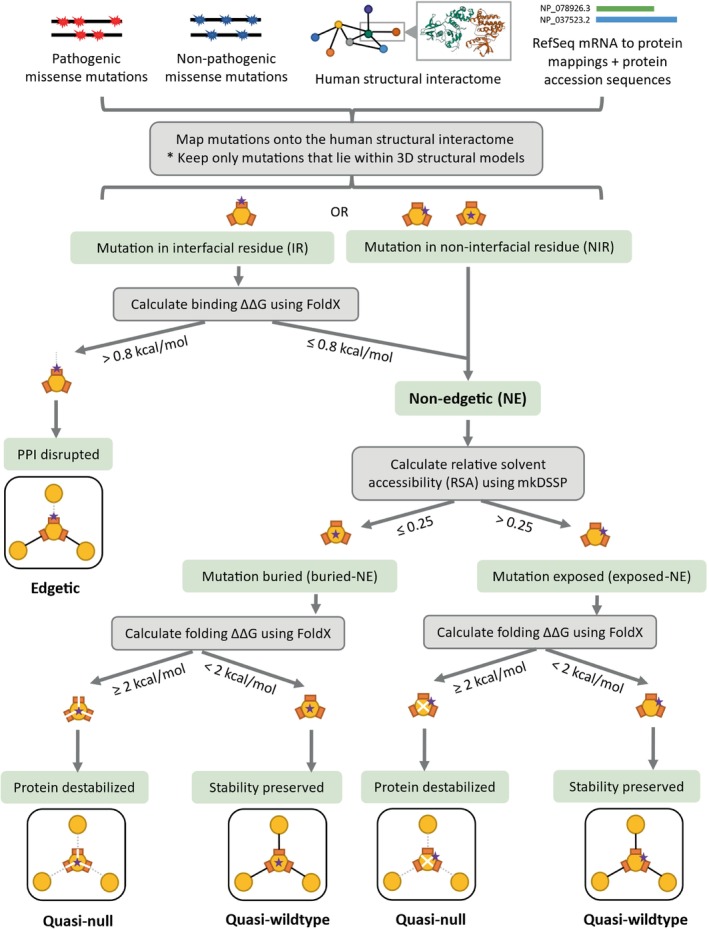
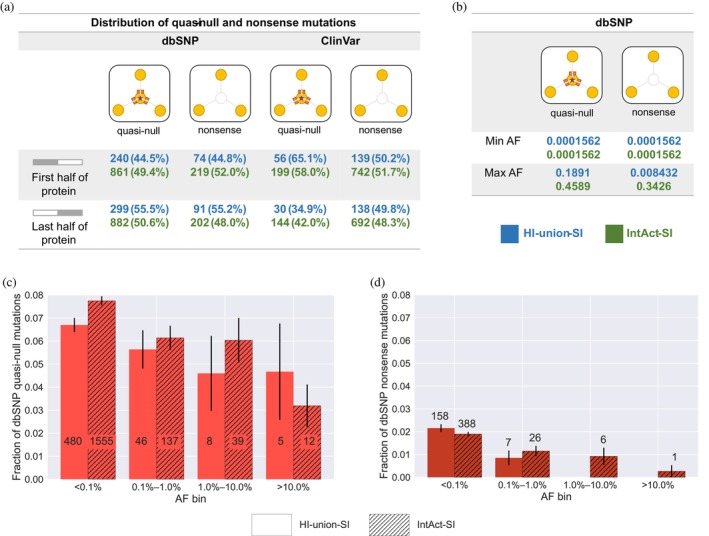
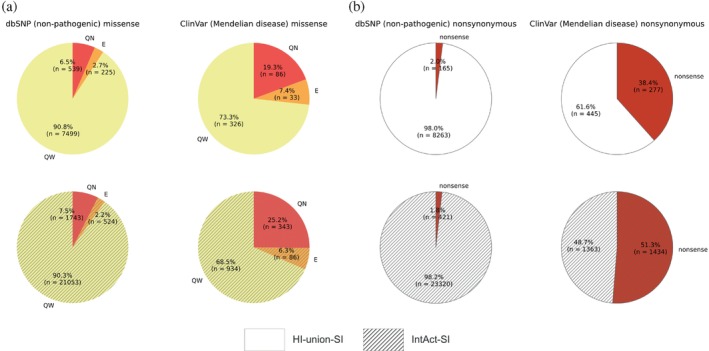
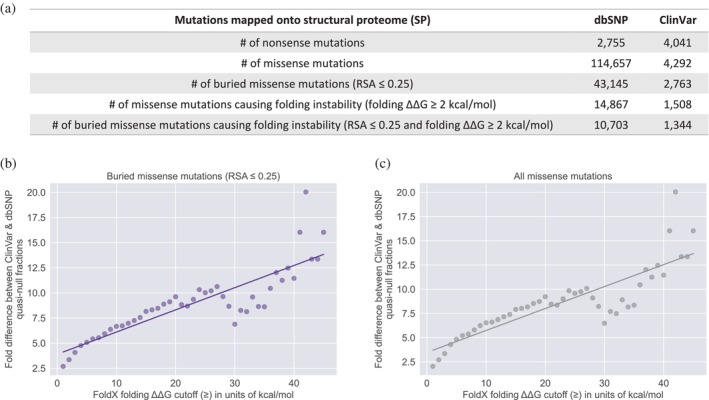
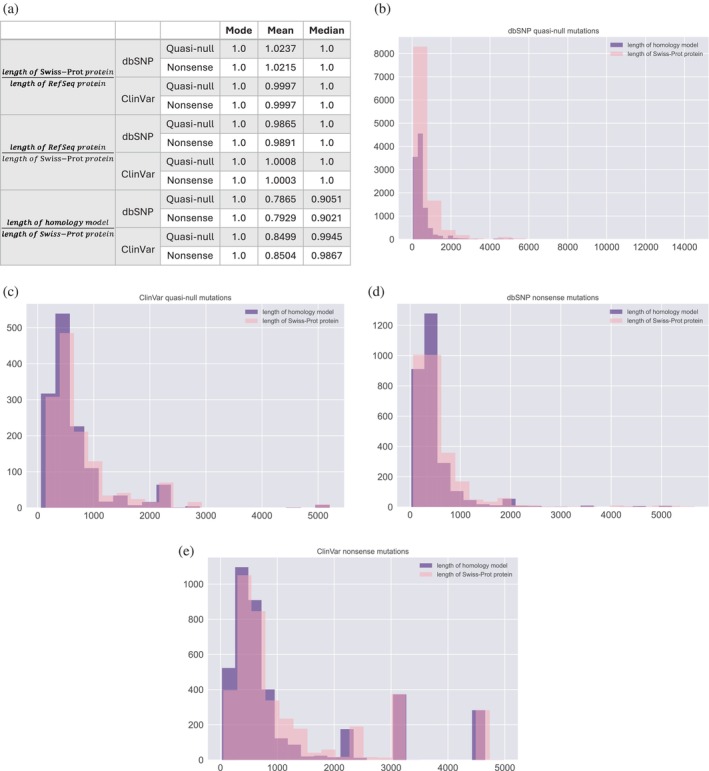
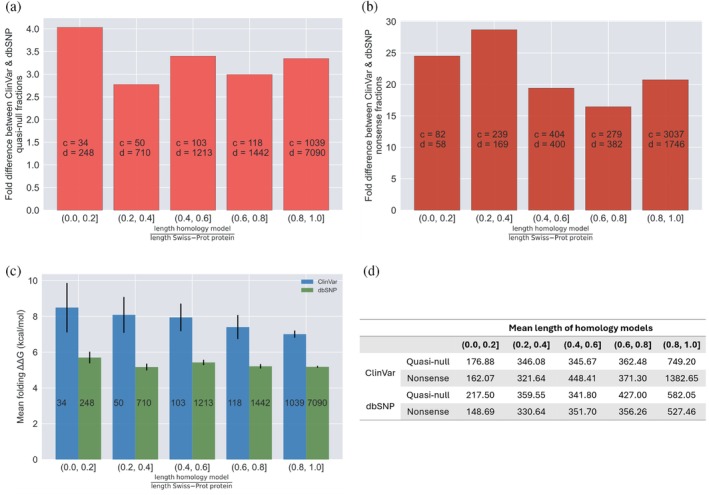
The complex genotype-to-phenotype relationships in Mendelian diseases can be elucidated by mutation-induced disturbances to the networks of molecular interactions (interactomes) in human cells. Missense and nonsense mutations cause distinct perturbations within the human protein interactome, leading to functional and phenotypic effects with varying degrees of severity. Here, we structurally resolve the human protein interactome at atomic-level resolutions and perform structural and thermodynamic calculations to assess the biophysical implications of these mutations. We focus on a specific type of missense mutation, known as "quasi-null" mutations, which destabilize proteins and cause similar functional consequences (node removal) to nonsense mutations. We propose a "fold difference" quantification of deleteriousness, which measures the ratio between the fractions of node-removal mutations in datasets of Mendelian disease-causing and non-pathogenic mutations. We estimate the fold differences of node-removal mutations to range from 3 (for quasi-null mutations with folding ΔΔG ≥2 kcal/mol) to 20 (for nonsense mutations). We observe a strong positive correlation between biophysical destabilization and phenotypic deleteriousness, demonstrating that the deleteriousness of quasi-null mutations spans a continuous spectrum, with nonsense mutations at the extreme (highly deleterious) end. Our findings substantiate the disparity in phenotypic severity between missense and nonsense mutations and suggest that mutation-induced protein destabilization is indicative of the phenotypic outcomes of missense mutations. Our analyses of node-removal mutations allow for the potential identification of proteins whose removal or destabilization lead to harmful phenotypes, enabling the development of targeted therapeutic approaches, and enhancing comprehension of the intricate mechanisms governing genotype-to-phenotype relationships in clinically relevant diseases.
Keywords: Mendelian diseases; deleteriousness of mutations; functional consequences; genotype‐to‐phenotype relationships; human genetic variants; human protein interactome; interactome disruptions; missense and nonsense mutations; protein stability; structural systems biology.
© 2025 The Author(s). Protein Science published by Wiley Periodicals LLC on behalf of The Protein Society.
Figures
Similar articles
-
Large scale analysis of protein stability in OMIM disease related human protein variants.BMC Genomics. 2016 Jun 23;17 Suppl 2(Suppl 2):397. doi: 10.1186/s12864-016-2726-y. BMC Genomics. 2016. PMID: 27356511 Free PMC article.
-
Differential functional readthrough over homozygous nonsense mutations contributes to the bleeding phenotype in coagulation factor VII deficiency.J Thromb Haemost. 2016 Oct;14(10):1994-2000. doi: 10.1111/jth.13443. Epub 2016 Sep 19. J Thromb Haemost. 2016. PMID: 27513915
-
Mutation Edgotype Drives Fitness Effect in Human.Front Bioinform. 2021 Aug 30;1:690769. doi: 10.3389/fbinf.2021.690769. eCollection 2021. Front Bioinform. 2021. PMID: 36303776 Free PMC article.
-
Can mucopolysaccharidosis type I disease severity be predicted based on a patient's genotype? A comprehensive review of the literature.Genet Med. 2003 Jul-Aug;5(4):286-94. doi: 10.1097/01.GIM.0000078027.83236.49. Genet Med. 2003. PMID: 12865757 Review.
-
Listening to silence and understanding nonsense: exonic mutations that affect splicing.Nat Rev Genet. 2002 Apr;3(4):285-98. doi: 10.1038/nrg775. Nat Rev Genet. 2002. PMID: 11967553 Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
