

Protein misfolding and mitochondrial dysfunction in glaucoma
- PMID: 40385286
- PMCID: PMC12083186
- DOI: 10.3389/fcell.2025.1595121
Protein misfolding and mitochondrial dysfunction in glaucoma
Abstract
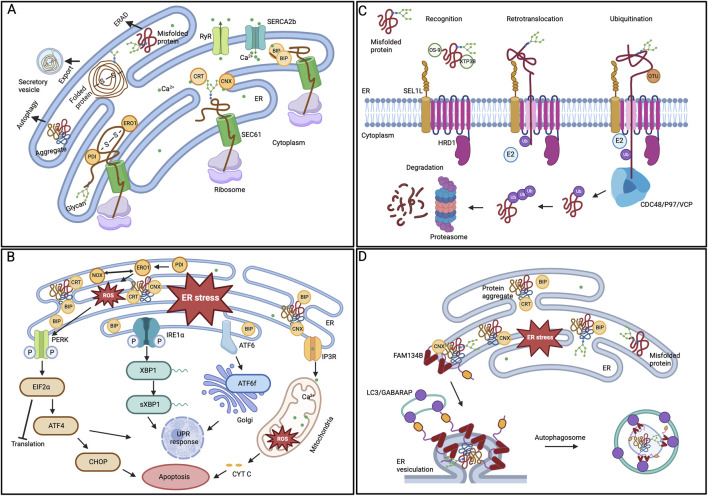
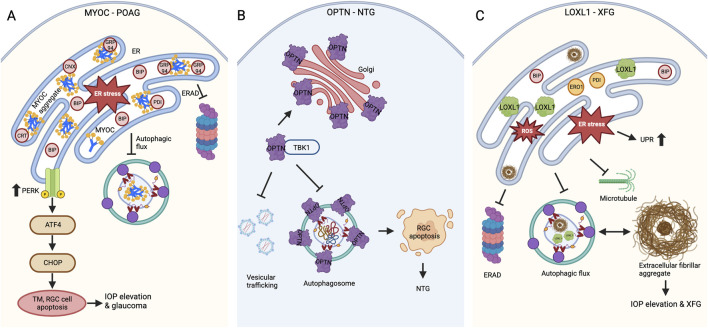
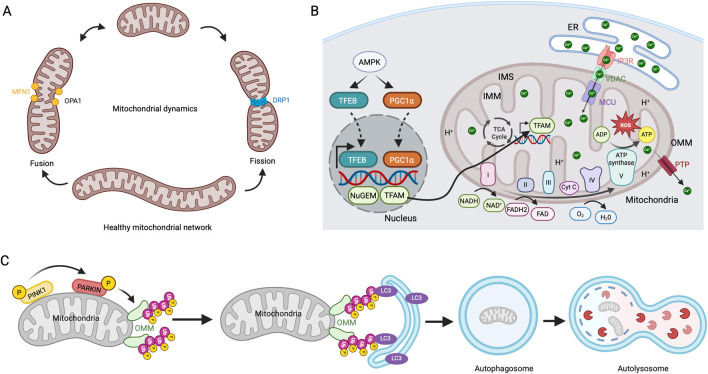
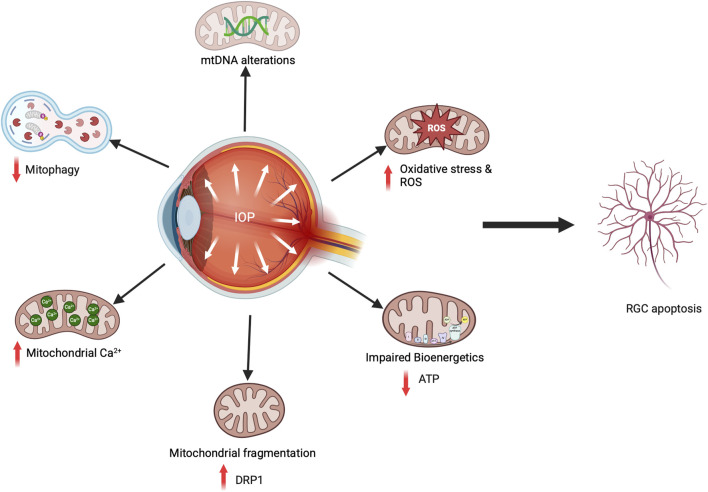
Glaucoma is a leading cause of irreversible blindness worldwide. Elevated intraocular pressure caused by restricted outflow of the aqueous humor leads to the degeneration of retinal ganglion cells (RGCs) and their axons. Emerging evidence suggests that pathological mechanisms relating to protein folding and mitochondrial dysfunction are significant factors in the disease onset of different types of open-angle glaucoma. In this review, we discuss these defects in three distinct types of open-angle glaucoma: primary open-angle glaucoma (POAG), normal tension glaucoma (NTG), and exfoliation glaucoma (XFG). Genetic mutations linked to the previously mentioned open-angle glaucoma, including those in myocilin (MYOC), optineurin (OPTN), and lysyl oxidase 1 (LOXL1), disrupt protein folding and homeostasis, leading to endoplasmic reticulum stress, activation of the unfolded protein response and impaired autophagic protein degradation. These factors contribute to trabecular meshwork and retinal ganglion cell apoptosis. In addition to protein folding defects, mitochondrial dysfunction is also associated with the progression of trabecular meshwork damage and the death of RGCs. Factors such as oxidative stress, an altered mitochondrial fission-fusion balance, and mitophagy dysregulation make RGCs vulnerable and contribute to optic nerve degeneration. The crosstalk between protein folding and mitochondrial defects in glaucoma underscores the complexity of disease pathogenesis and offers potential targets for therapeutic intervention. Strategies aimed at restoring protein homeostasis, enhancing mitochondrial function, and mitigating cellular stress responses hold promise for neuroprotection in glaucoma.
Keywords: NTG; POAG; XFG; autophagy; er stress; glaucoma; mitochondrial dysfunction; upr.
Copyright © 2025 Venkatesan and Bernstein.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Adam M. F., Belmouden A., Binisti P., Brezin A. P., Valtot F., Bechetoille A., et al. (1997). Recurrent mutations in a single exon encoding the evolutionarily conserved olfactomedin-homology domain of TIGR in familial open-angle glaucoma. Hum. Mol. Genet. 6, 2091–2097. 10.1093/hmg/6.12.2091 - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources