

This is a preprint.
Pathogenic variants in BORCS5 Cause a Spectrum of Neurodevelopmental and Neurodegenerative Disorders with Lysosomal Dysfunction
- PMID: 40385417
- PMCID: PMC12083638
- DOI: 10.1101/2025.04.30.25326597
Pathogenic variants in BORCS5 Cause a Spectrum of Neurodevelopmental and Neurodegenerative Disorders with Lysosomal Dysfunction
Abstract
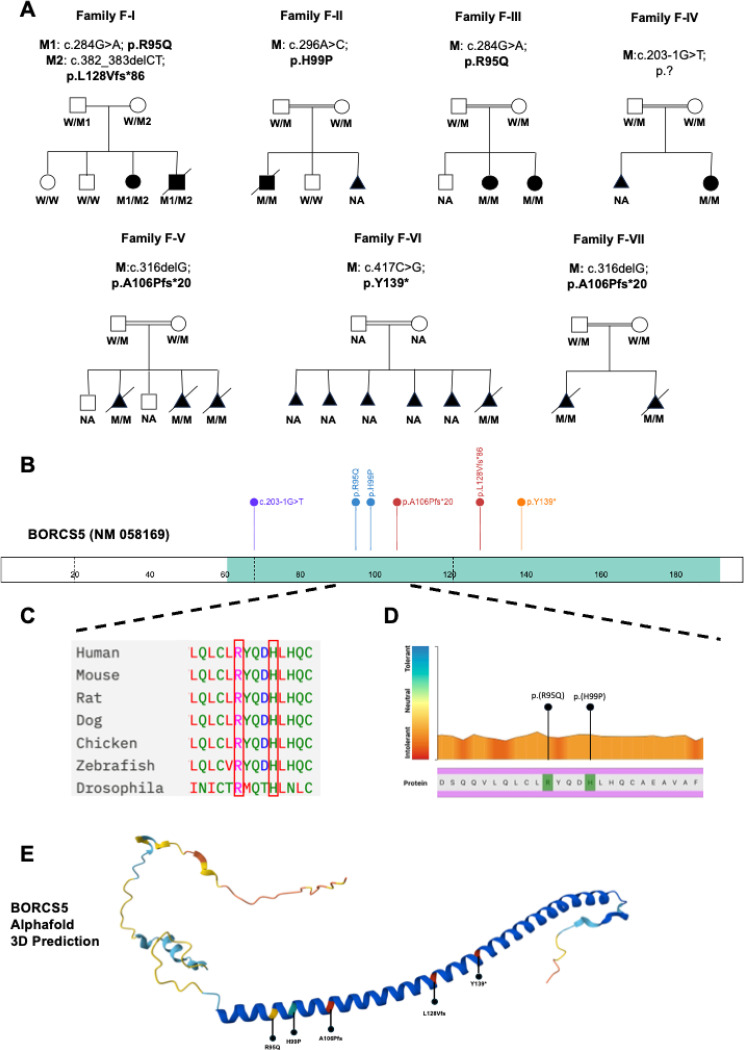
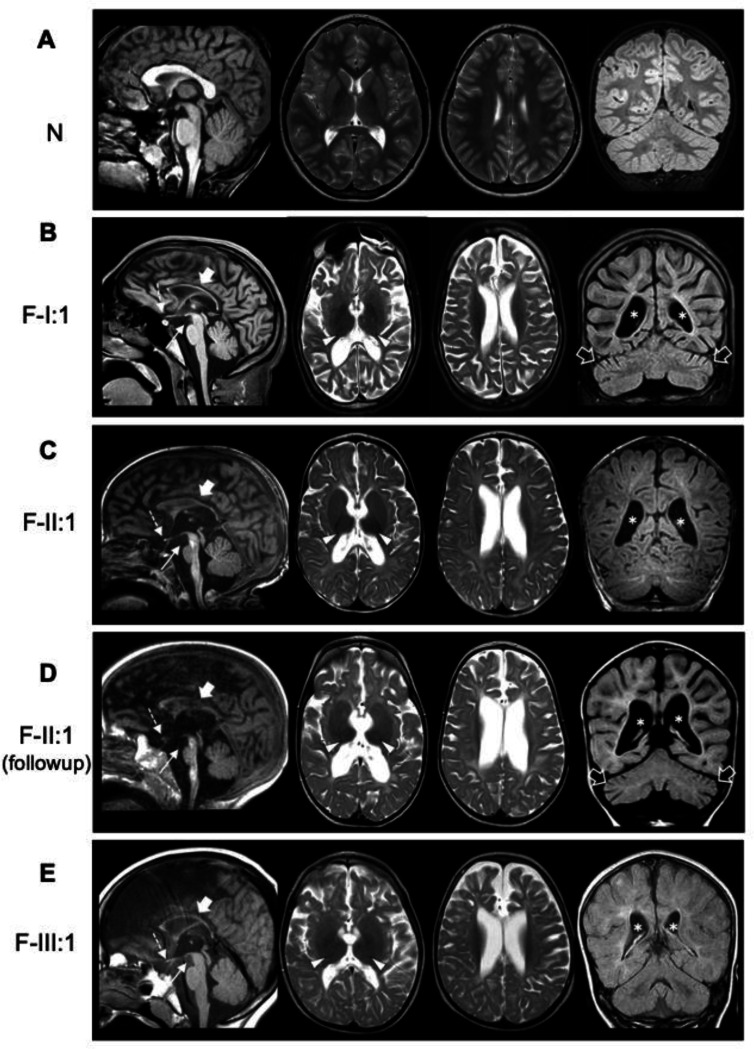
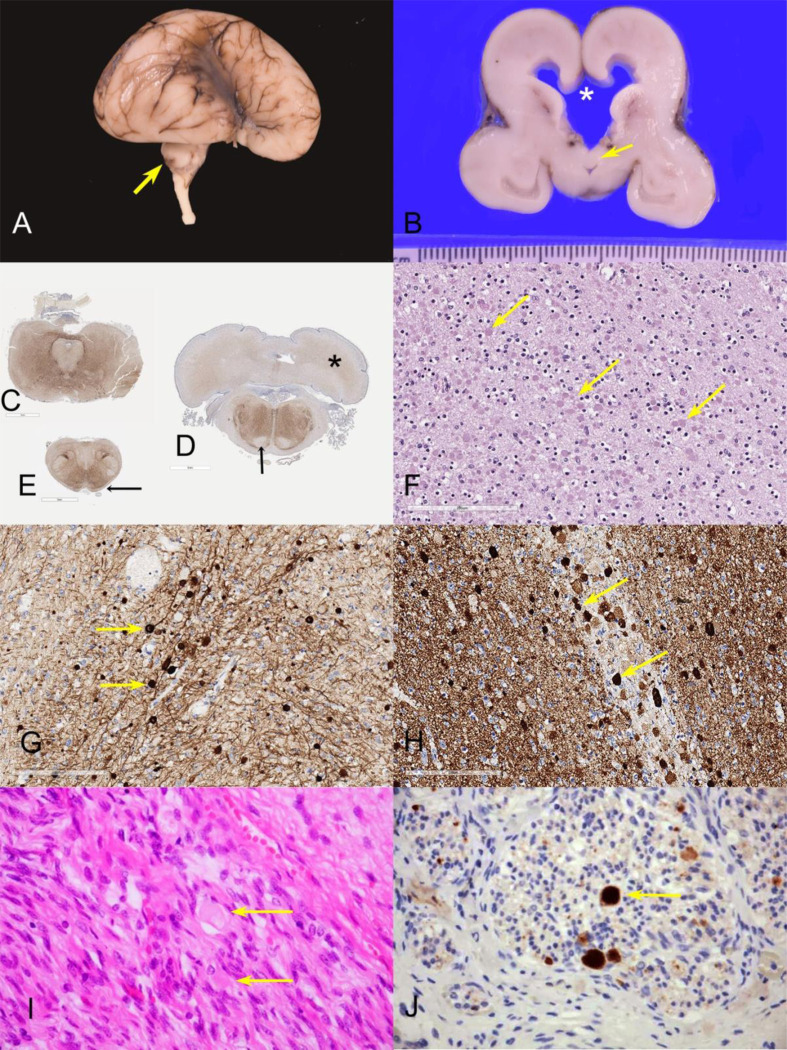
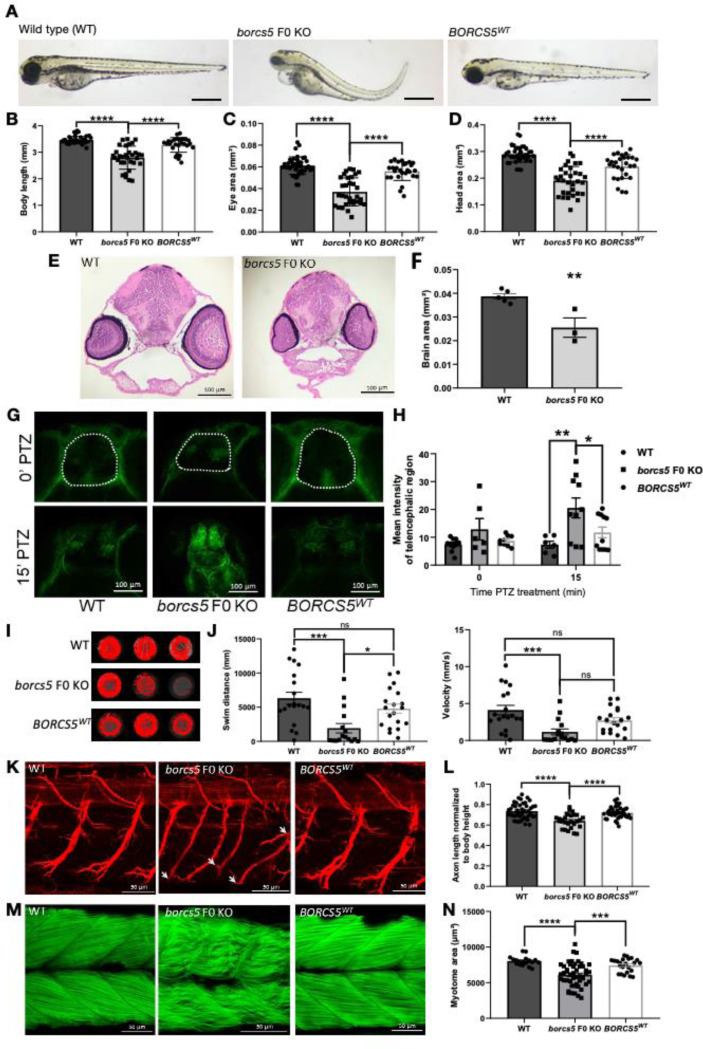
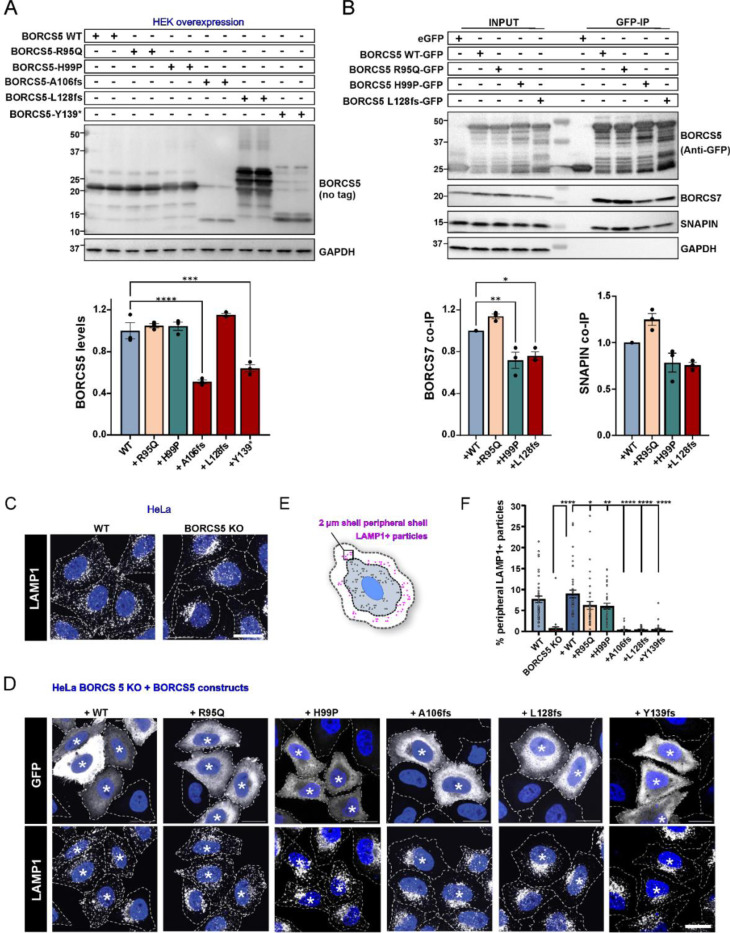
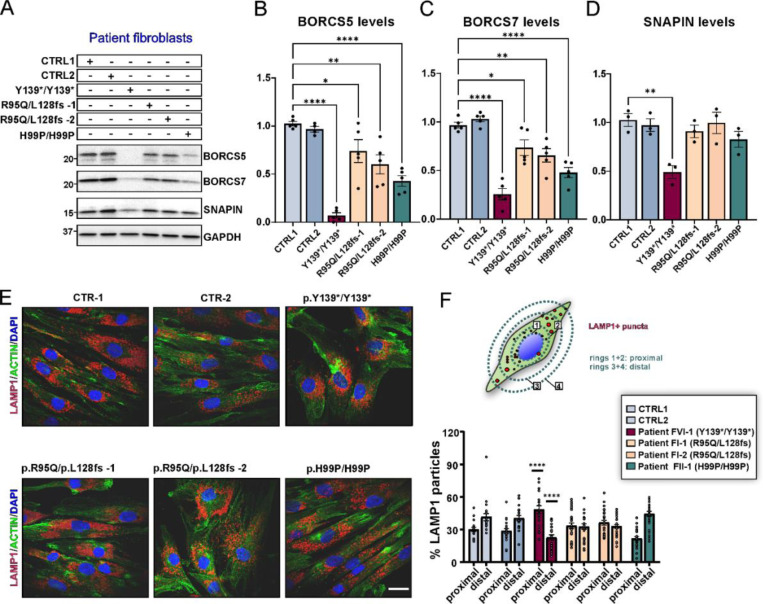
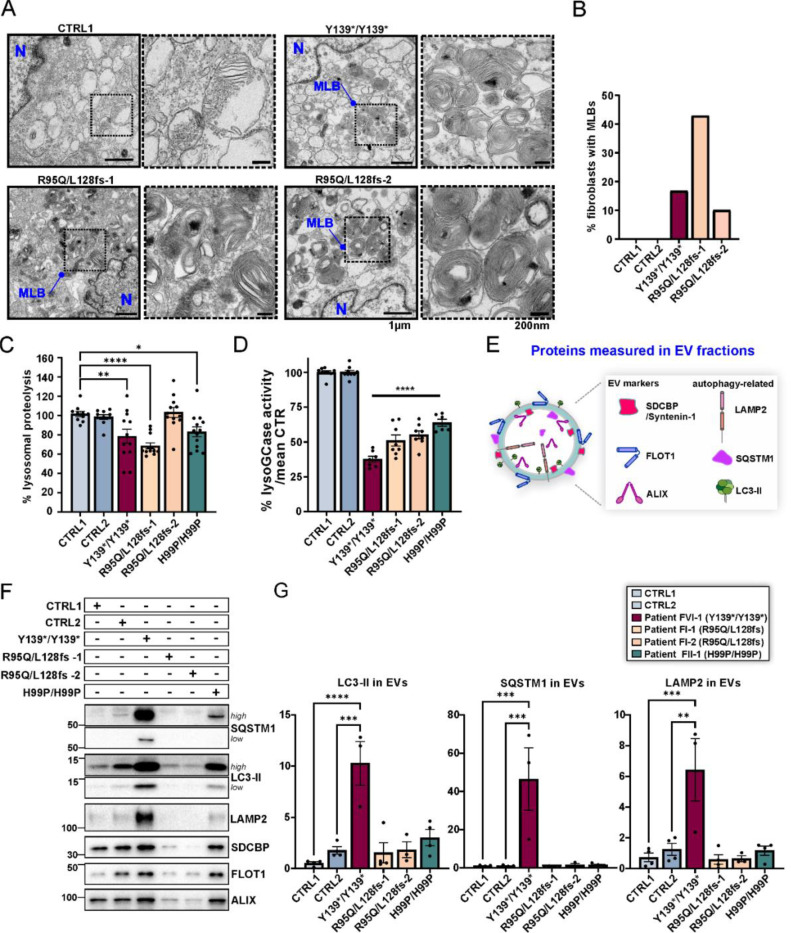
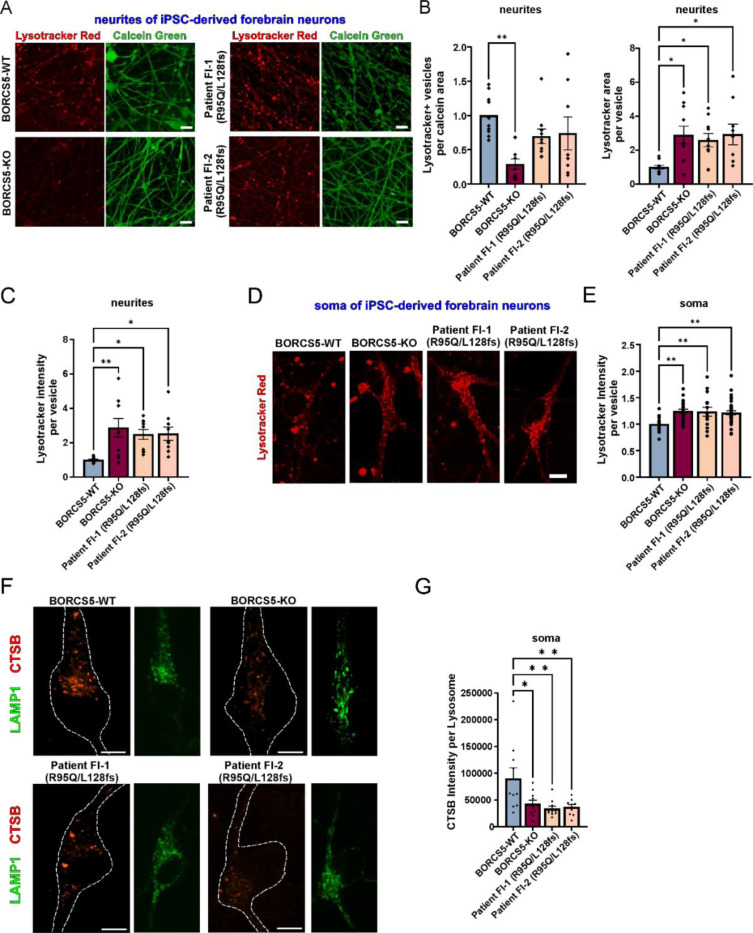
BORCS5 encodes a subunit of the BLOC-one-related complex (BORC), which is known to mediate the kinesin-dependent anterograde movement of lysosomes. Using whole-exome sequencing, we identified 12 cases from seven families carrying bi-allelic BORCS5 variants, including four loss-of-function and two missense variants. Carriers of homozygous loss-of-function variants presented with prenatally lethal arthrogryposis multiplex congenita, brain malformations, and neuropathological evidence of diffuse neuroaxonal dystrophy. Individuals with missense variants presented differently, with microcephaly, developmental epileptic encephalopathy, intellectual disability, optic atrophy, spasticity, and progressive movement disorders. In this group, brain MRI showed diffuse hypomyelination and progressive global cerebral atrophy, consistent with neurodegeneration. Borcs5 knockout in zebrafish exhibited microcephaly, motor deficits, and seizures, mirroring the patients' clinical presentation. At the cellular level, BORCS5 loss-of-function but not missense variants, resulted in lower protein expression and impaired BORC assembly, paralleled by perinuclear lysosomal clustering. However, both loss-of-function and missense BORCS5 variants were associated with reduced total lysosomal proteolysis, reduced activity of the lysosomal hydrolases glucocerebrosidase and cathepsin B, and presence of multilamellar bodies, indicating lysosomal dysfunction. Our study reveals a novel role for BORCS5 in the regulation of lysosomal function, in addition to its known role in the anterograde movement of lysosomes, possibly underlying the diverse clinical manifestations in individuals with BORCS5-related disorders.
Conflict of interest statement
Niccolò E. Mencacci receives NIH funding (1K08NS131581) and is supported by the Align Science Across Parkinson’s (ASAP) Global Parkinson’s Genetics Program (GP2). He is a member of the steering committee of the PD GENEration study for which he receives an honorarium from the Parkinson’s Foundation. Francesca Magrinelli is supported by the NIHR UCLH Biomedical Research Centre (BRC) Translational Neuroscience Intermediate Clinical Fellowship (Grant ID: BRC1287/TN/FM/101410), the Edmond J. Safra Movement Disorders Research Career Development Award (Grant ID: MJFF-023893), Parkinson’s UK (Grant ID: G-2401), the American Parkinson Disease Association (Grant ID: 1282403) and the David Pearlman Charitable Foundation. Ingrid M Wentzensen is an employee of and may own stock in GeneDx, LLC. Kailash Bhatia has received grant support from the Edmond J. Safra Movement Disorders Research Career Development Award (Grant ID: MJFF-023893), Parkinson’s UK (Grant ID: G-2401), EPSRC, and the David Pearlman Charitable Foundation. He has received a stipend from the international Parkinson’s disease and Movement Disorders Society as editor of MDCP journal and book royalties from Oxford University Press. Dimitri Krainc is the Founder and Scientific Advisory Board Chair of Lysosomal Therapeutics Inc. and Vanqua Bio. He also serves on the scientific advisory boards of The Silverstein Foundation, Intellia Therapeutics, AcureX, and Prevail Therapeutics and is a Venture Partner at OrbiMed.
Figures
References
-
- Steel D. et al. Loss-of-Function Variants in HOPS Complex Genes VPS16 and VPS41 Cause Early Onset Dystonia Associated with Lysosomal Abnormalities. Ann Neurol 88, 867–877 (2020). - PubMed
-
- Ballabio A. & Bonifacino J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol 21, 101–118 (2020). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources