

Primary Lateral Sclerosis: Implications for Diagnostic Criteria From a Natural History Study in the Netherlands
- PMID: 40388677
- PMCID: PMC12092547
- DOI: 10.1212/WNL.0000000000213461
Primary Lateral Sclerosis: Implications for Diagnostic Criteria From a Natural History Study in the Netherlands
Abstract
Background and objectives: Primary lateral sclerosis (PLS) is a rare disease characterized by upper motor neuron (UMN) degeneration. We aimed to elucidate the natural history in patients with UMN syndromes suggestive of PLS and validate the most recent diagnostic (consensus) criteria.
Methods: A validation study of a long-term follow-up cohort was conducted, including adults with UMN syndromes and disease durations ≥6 months. Patients were assessed at baseline (T1), at 3 years (T2), and when possible after 13 years (T3). Diagnostic categorization followed the 2020 PLS consensus criteria. Main outcomes included diagnostic classification at follow-up and survival.
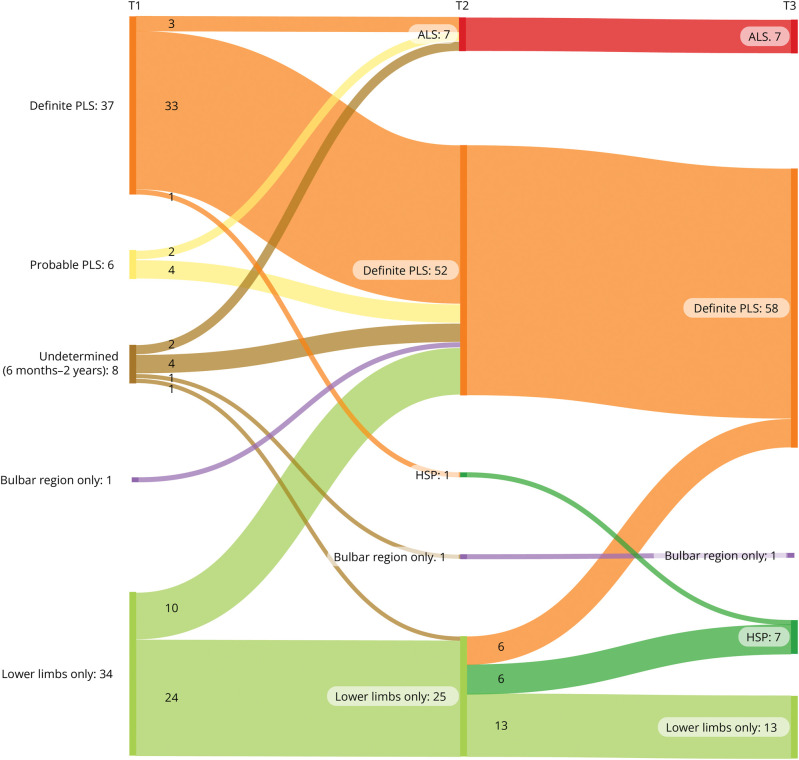
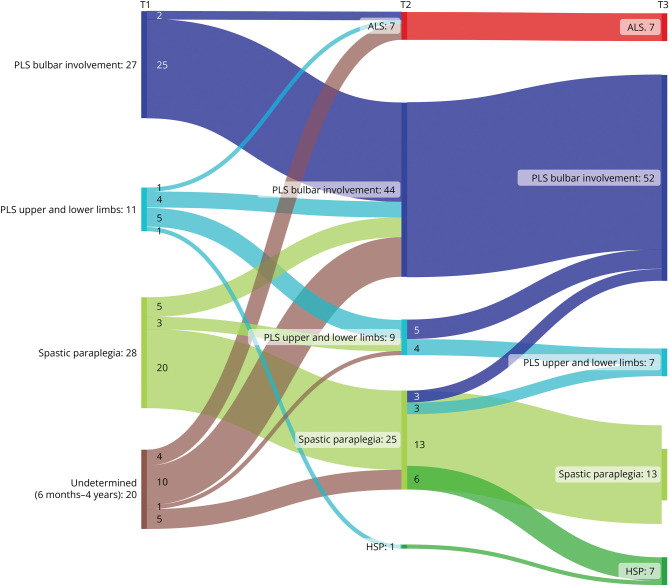
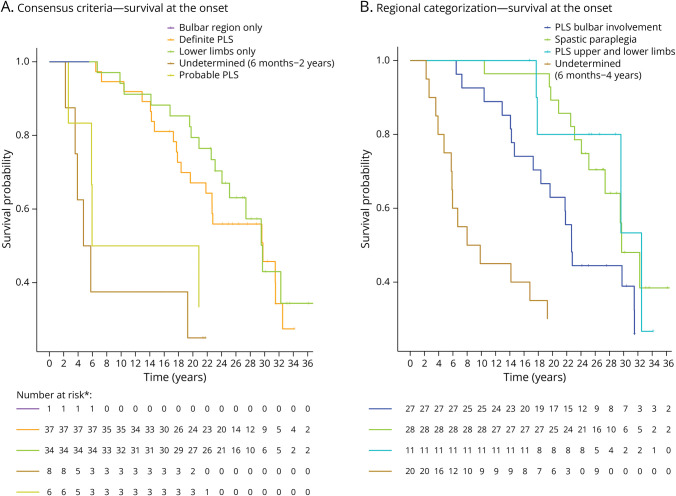
Results: The study comprised 86 patients (34 women [40%], mean age 58.9 ± 10.1 years), of whom 43 met the PLS consensus criteria at baseline (6 probable, 37 definite). Eight patients had a disease duration <2 years, and 35 patients presented with UMN symptoms localized to 1 region (1 bulbar, 34 legs). Change of initial diagnosis occurred in 14% of patients with PLS, and 49% of patients presenting with UMN symptoms in 1 region progressed to PLS. Seven patients developed amyotrophic lateral sclerosis (ALS), and for 7 patients, diagnosis was revised to hereditary spastic paraplegia (HSP). Survival was shorter for patients with a disease duration <4 years. In the probable PLS group, 33% converted to ALS. Converters had a steeper Amyotrophic Lateral Sclerosis Functional Rating Scale slope (p = 0.023) and shorter symptom duration (p < 0.001) at inclusion. Of patients presenting with leg symptoms, diagnosis was revised between T2 and T3 in 29%. Introducing a 4-year minimal disease duration for PLS diagnosis and categorization based on regions involved resulted in 86% of PLS diagnoses remaining within the PLS category, 5% transitioning to ALS (slow variant), and 9% to HSP. Survival was longest for patients presenting with symptoms confined to arms and legs or legs only, followed by those with bulbar involvement at baseline, while patients with disease durations between 6 months and 4 years exhibited the shortest survival.
Discussion: Our findings suggest that a diagnosis of PLS should be deferred until 4 years after symptom onset because shorter durations correlate with higher ALS conversion rates and shorter survival. Categorization by regional involvement may facilitate more effective monitoring of patients with UMN syndromes.
Conflict of interest statement
B.S. de Vries, E.M.J. de Boer, F. Brugman, and P. Van Damme report no disclosures relevant to the manuscript. J.H. Veldink reports sponsored research agreements with Biogen and Astra Zeneca. M.A. van Es has consulted for Biogen; has received travel grants from Shire (formerly Baxalta); has performed work as a medical monitor for Ferrer (NCT05178810) with fees going to his institution; receives funding support from the Netherlands Organization for Health Research and Development (Vidi scheme), The Thierry Latran Foundation, the Motor Neurone Disease Association, FIGHT-MND, and the ALS Foundation Netherlands; is a member of the European Reference Network for Neuromuscular Diseases (EURO-NMD); and serves as a board member for the Spierziekten Centrum Nederland (SCN). L.H. van den Berg declares fees to his institution from Biogen, Wave, Amylyx, Ferrer, and Cytokinetics for being on scientific advisory boards; fees to his institution from Amylyx for a lecture; and an unrestricted educational grant from Takeda; and is the chair of ENCALS and TRICALS. Go to
Figures
Comment in
-
Primary Lateral Sclerosis (Progressive Spinobulbar Spasticity): Serial Analysis Over Many Years Increases Diagnostic Certainty.Neurology. 2025 Jun 10;104(11):e213685. doi: 10.1212/WNL.0000000000213685. Epub 2025 May 19. Neurology. 2025. PMID: 40388676 No abstract available.
Similar articles
-
Iron-sensitive MR imaging of the primary motor cortex to differentiate hereditary spastic paraplegia from other motor neuron diseases.Eur Radiol. 2022 Dec;32(12):8058-8064. doi: 10.1007/s00330-022-08865-6. Epub 2022 May 20. Eur Radiol. 2022. PMID: 35593959
-
Cerebrospinal Fluid Neurofilaments May Discriminate Upper Motor Neuron Syndromes: A Pilot Study.Neurodegener Dis. 2018;18(5-6):255-261. doi: 10.1159/000493986. Epub 2018 Nov 14. Neurodegener Dis. 2018. PMID: 30428468
-
Upper motor neuron signs in primary lateral sclerosis and hereditary spastic paraplegia.Muscle Nerve. 2024 Jul;70(1):152-156. doi: 10.1002/mus.28100. Epub 2024 Apr 30. Muscle Nerve. 2024. PMID: 38687249
-
Demographics and clinical characteristics of primary lateral sclerosis: case series and a review of literature.Neurodegener Dis Manag. 2018 Feb;8(1):17-23. doi: 10.2217/nmt-2017-0051. Epub 2018 Jan 10. Neurodegener Dis Manag. 2018. PMID: 29316850 Review.
-
Could PLS represent a UMN-predominant ALS syndrome?Rev Neurol (Paris). 2025 Jan-Feb;181(1-2):52-57. doi: 10.1016/j.neurol.2024.04.006. Epub 2024 May 22. Rev Neurol (Paris). 2025. PMID: 38782644 Review.
References
-
- Corcia P, Honnorat J, Guennoc AM, de Toffol B, Autret A. Primary lateral sclerosis with breast cancer, a potential paraneoplastic neurological syndrome [in French]. Rev Neurol (Paris). 2000;156(11):1020-1022. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous