

DNA replication timing reveals genome-wide features of transcription and fragility
- PMID: 40389432
- PMCID: PMC12089344
- DOI: 10.1038/s41467-025-59991-w
DNA replication timing reveals genome-wide features of transcription and fragility
Abstract
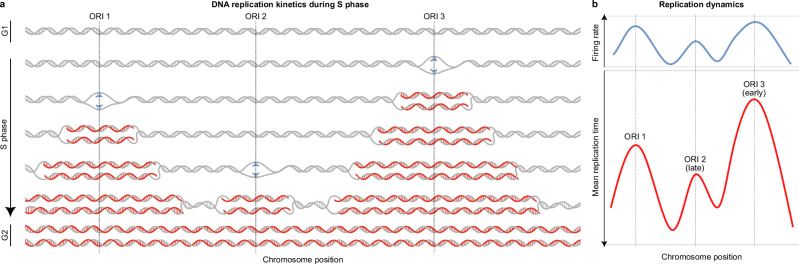
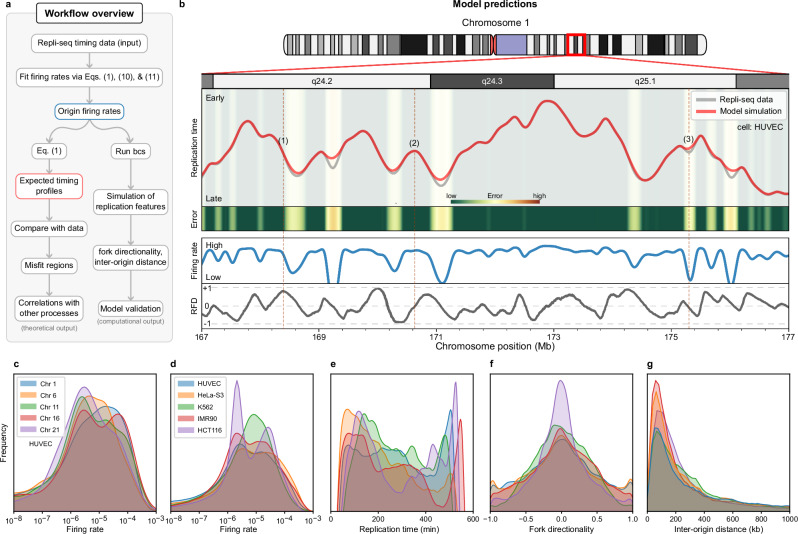
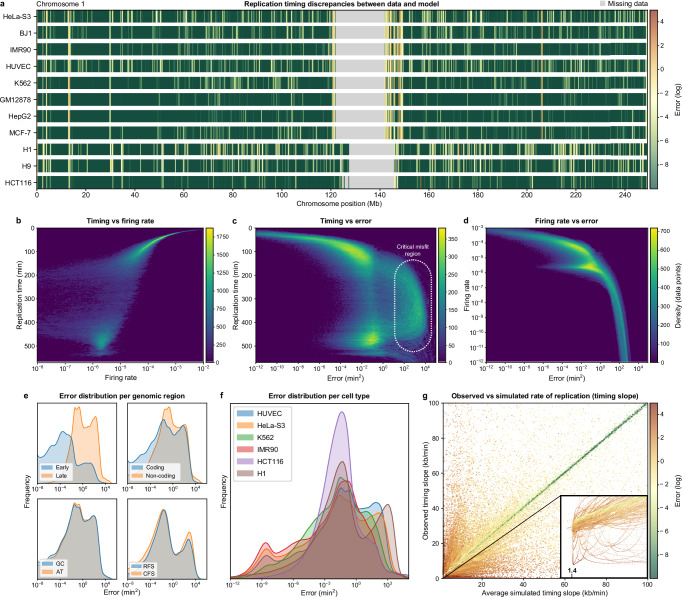
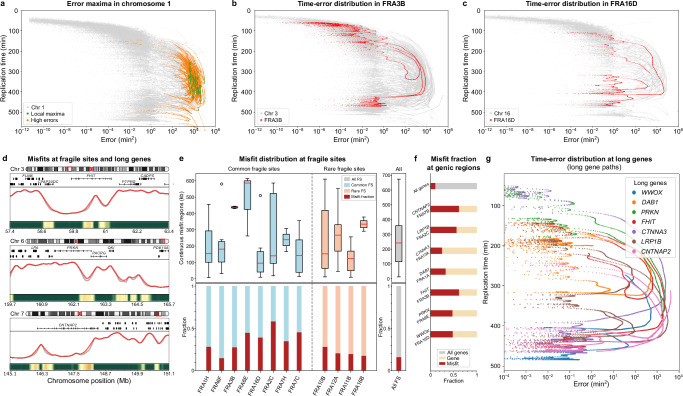
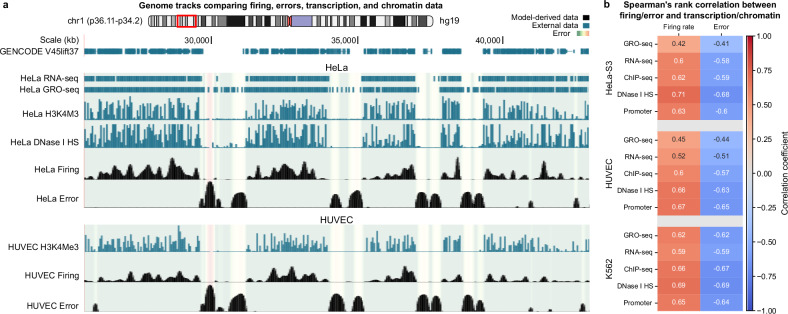
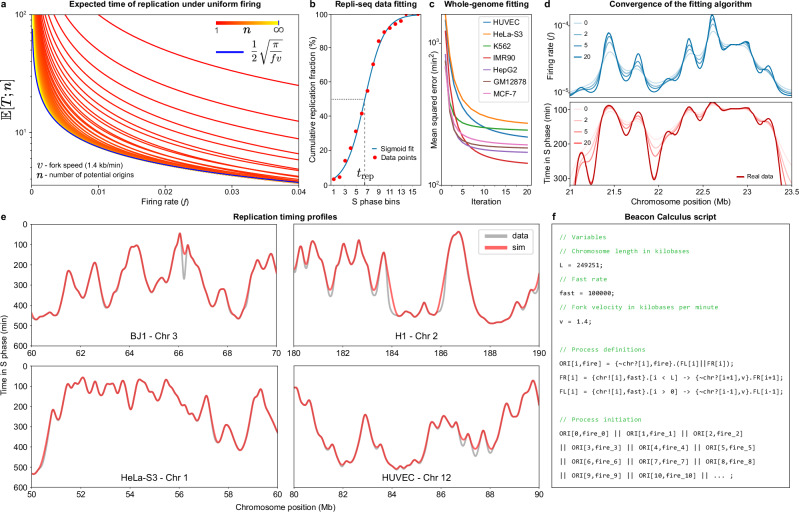
DNA replication in humans requires precise regulation to ensure accurate genome duplication and maintain genome integrity. A key indicator of this regulation is replication timing, which reflects the interplay between origin firing and fork dynamics. We present a high-resolution (1-kilobase) mathematical model that infers firing rate distributions from Repli-seq timing data across multiple cell lines, enabling a genome-wide comparison between predicted and observed replication. Notably, regions where the model and data diverge often overlap fragile sites and long genes, highlighting the influence of genomic architecture on replication dynamics. Conversely, regions of strong concordance are associated with open chromatin and active promoters, where elevated firing rates facilitate timely fork progression and reduce replication stress. In this work, we provide a valuable framework for exploring the structural interplay between replication timing, transcription, and chromatin organisation, offering insights into the mechanisms underlying replication stress and its implications for genome stability and disease.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
3D genome organization contributes to genome instability at fragile sites.Nat Commun. 2020 Jul 17;11(1):3613. doi: 10.1038/s41467-020-17448-2. Nat Commun. 2020. PMID: 32680994 Free PMC article.
-
Transcription-mediated organization of the replication initiation program across large genes sets common fragile sites genome-wide.Nat Commun. 2019 Dec 13;10(1):5693. doi: 10.1038/s41467-019-13674-5. Nat Commun. 2019. PMID: 31836700 Free PMC article.
-
A chromatin structure-based model accurately predicts DNA replication timing in human cells.Mol Syst Biol. 2014 Mar 28;10(3):722. doi: 10.1002/msb.134859. Mol Syst Biol. 2014. PMID: 24682507 Free PMC article.
-
[Regulation of DNA replication timing].Mol Biol (Mosk). 2013 Jan-Feb;47(1):12-37. doi: 10.1134/s0026893312060118. Mol Biol (Mosk). 2013. PMID: 23705493 Review. Russian.
-
Behavior of replication origins in Eukaryota - spatio-temporal dynamics of licensing and firing.Cell Cycle. 2015;14(14):2251-64. doi: 10.1080/15384101.2015.1056421. Epub 2015 Jun 1. Cell Cycle. 2015. PMID: 26030591 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
