

Mutations in NEK1 cause ciliary dysfunction as a novel pathogenic mechanism in amyotrophic lateral sclerosis
- PMID: 40389989
- PMCID: PMC12090460
- DOI: 10.1186/s13024-025-00848-7
Mutations in NEK1 cause ciliary dysfunction as a novel pathogenic mechanism in amyotrophic lateral sclerosis
Abstract
Background: Neuronal primary cilia, vital for signaling and cell-cycle regulation, have been implicated in maintaining neuronal identity. While a link between primary ciliary defects and neurodegenerative diseases is emerging, the precise pathological mechanisms remain unclear.
Methods: We studied the genetic contribution of NEK1 to ALS pathogenesis by analyzing the exome sequences of 920 Korean patients with ALS. To understand the disease contribution of NEK1 variants in ALS, we performed a series of functional studies using patient fibroblasts focusing on primary cilia and microtubule-related phenotypes. In addition, these findings were validated in iPSC-derived motor neurons (iPSC-MNs).
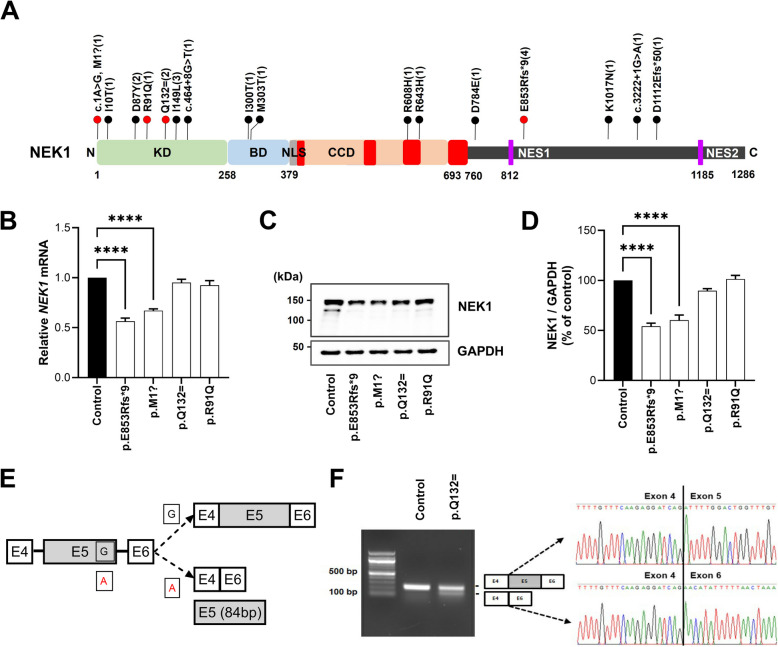
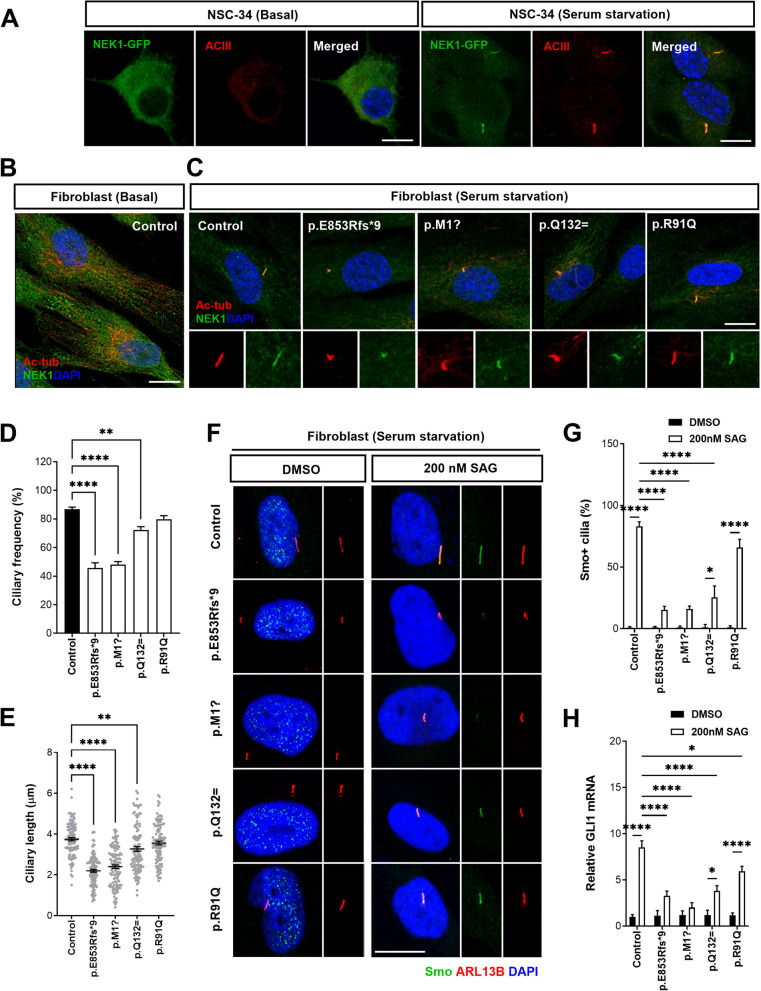
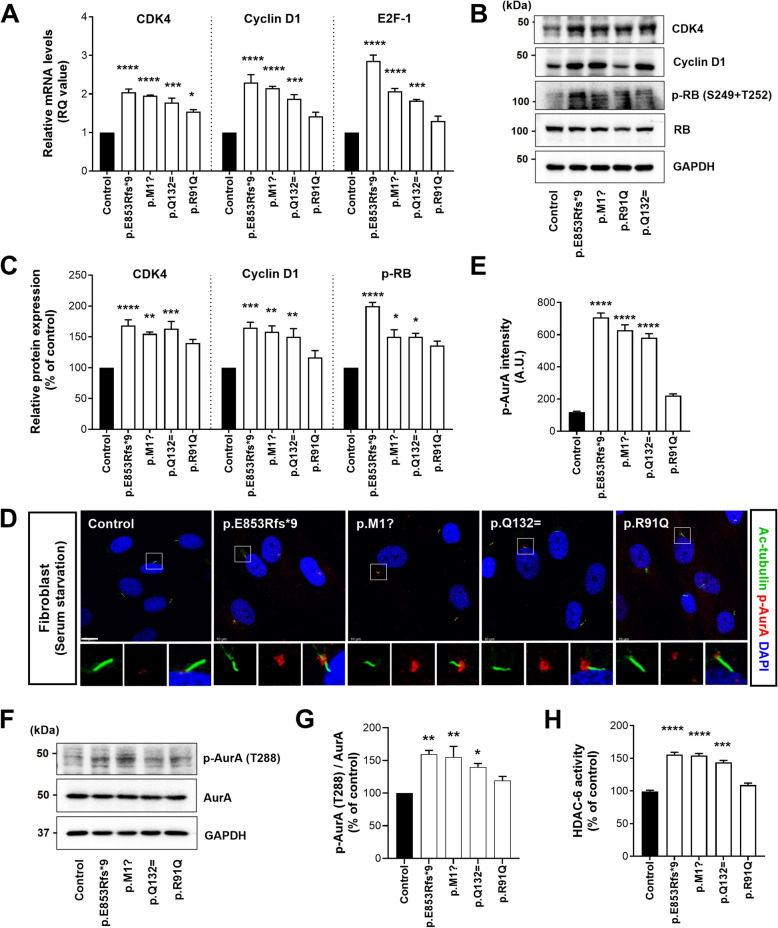
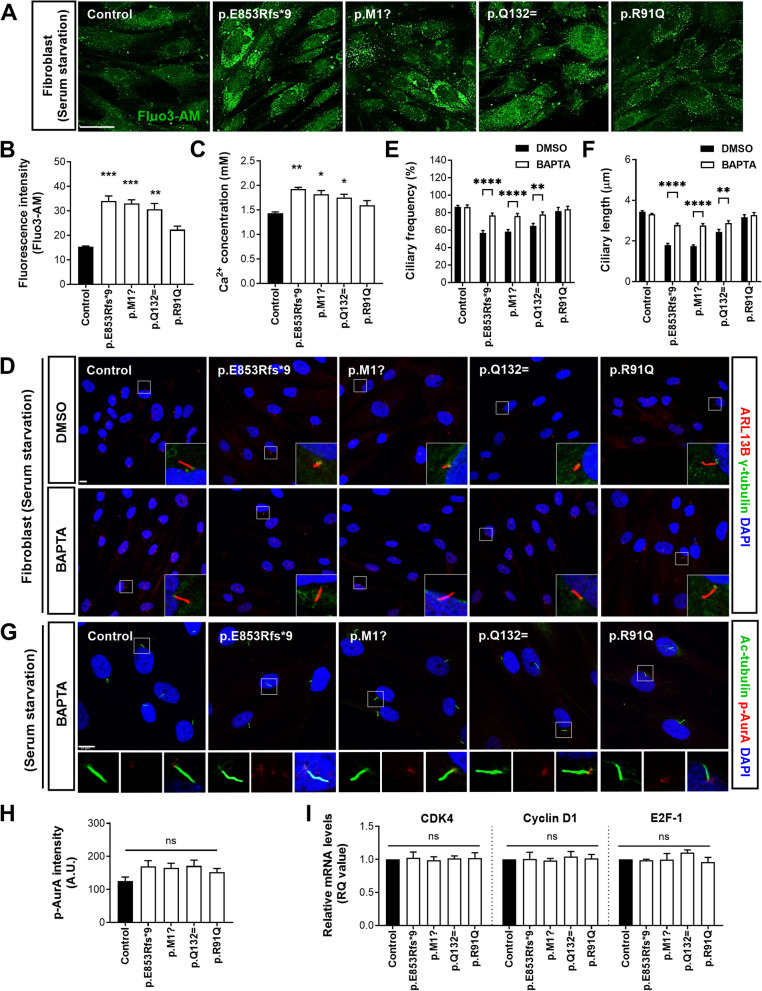
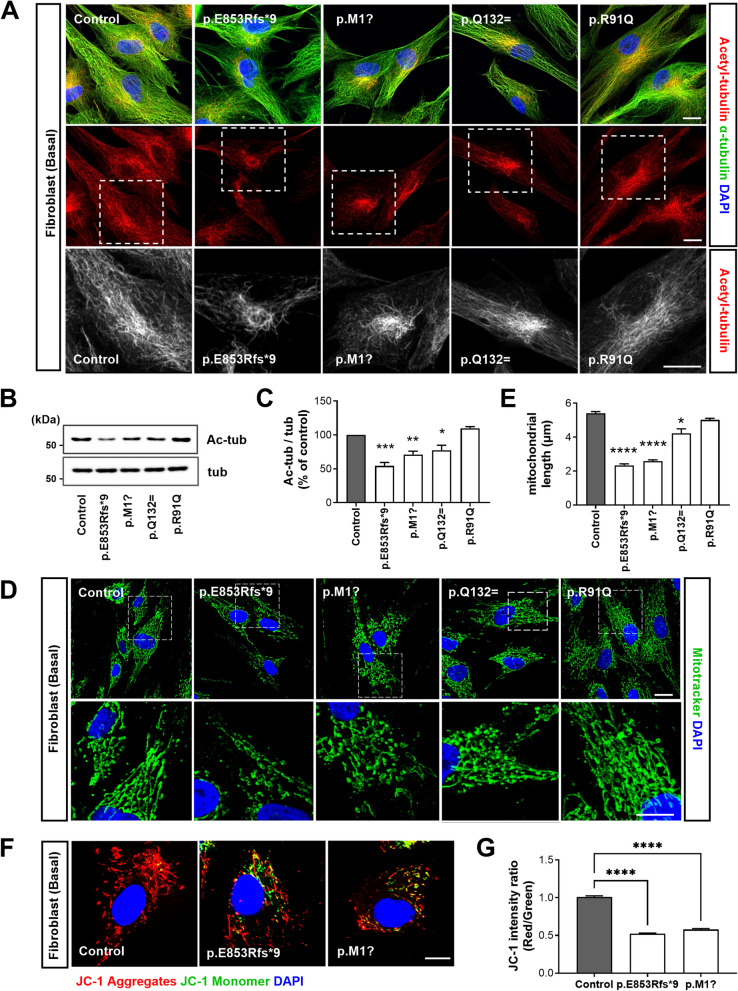
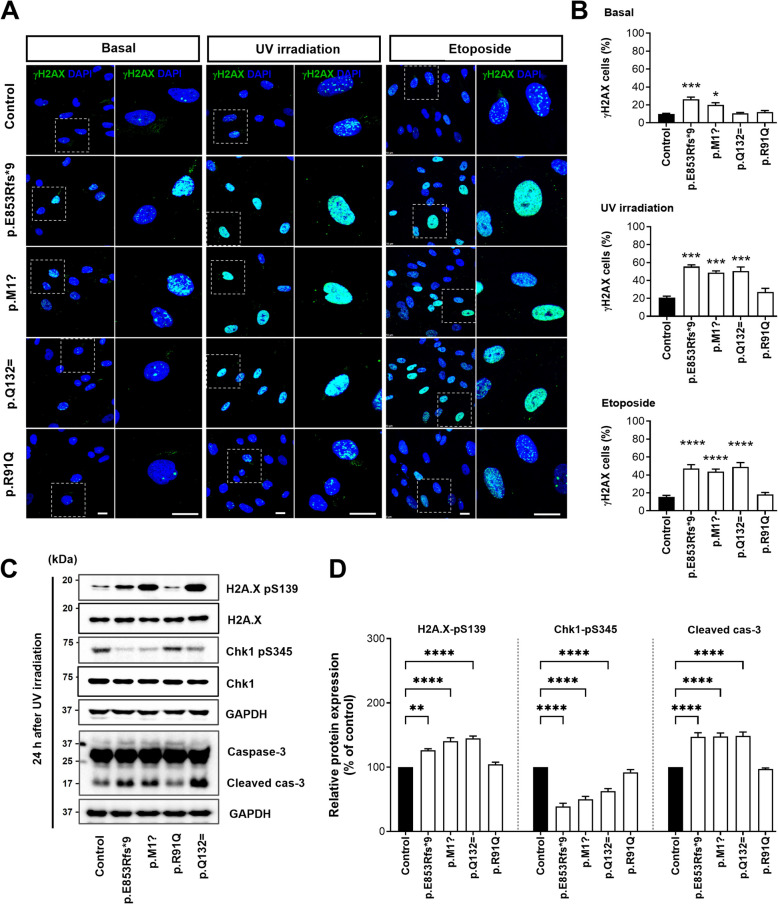
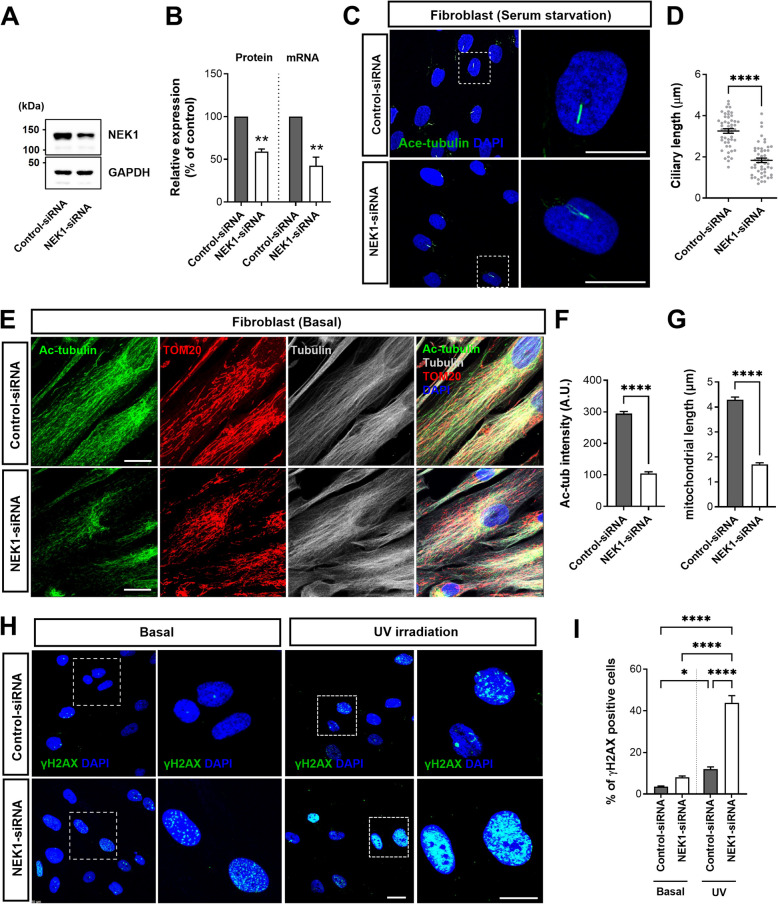
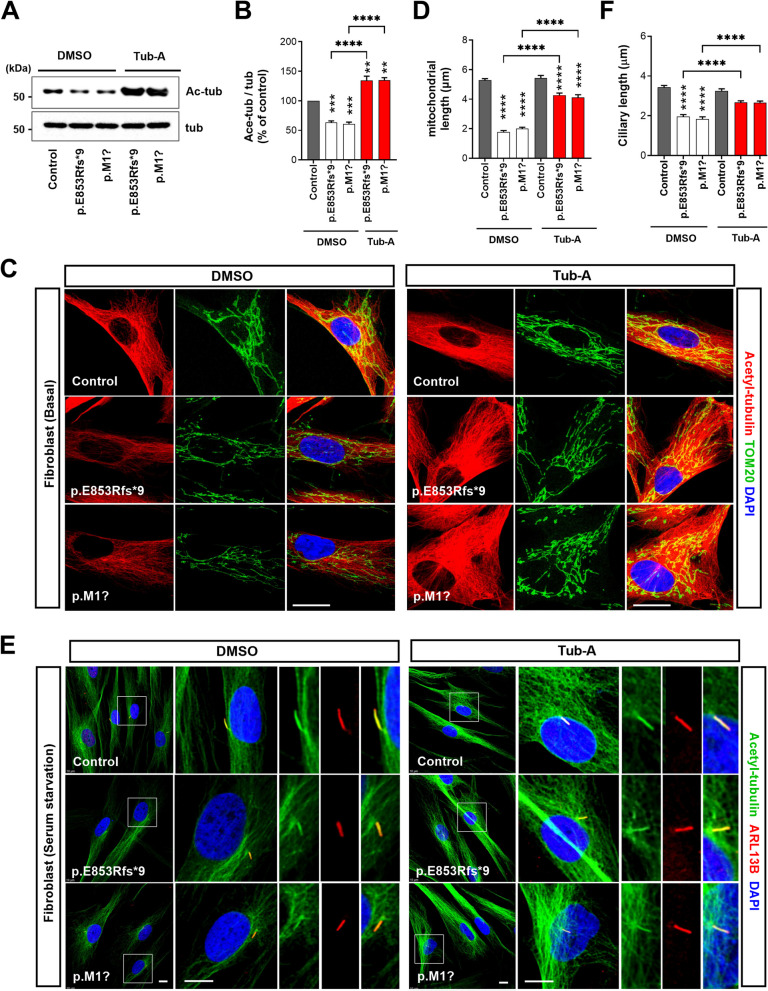
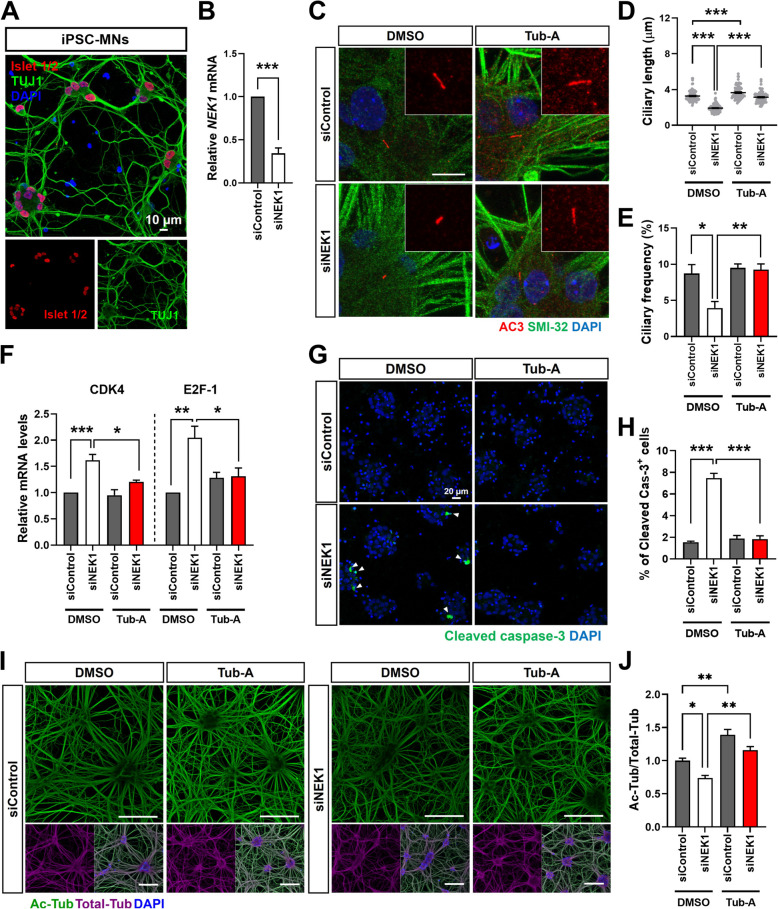
Results: NIMA-related kinase 1 (NEK1), a gene encoding a serine/threonine kinase involved in cell cycle regulation, has been identified as a risk gene for amyotrophic lateral sclerosis (ALS). Here, we report that mutations in NEK1 cause primary ciliary abnormality, cell cycle re-entry, and disrupted tubulin acetylation in ALS. We analyzed the whole-exome sequences of 920 Korean patients with sporadic ALS and identified 16 NEK1 variants in 23 patients. We found that two novel variants, p.E853Rfs*9 and p.M1?, reduced NEK1 expression, resulting in loss-of-function (LOF) and one synonymous splicing variant (p.Q132=) exhibited an aberrant isoform lacking exon 5. All three NEK1 variants exhibited abnormal primary ciliary structure, impaired sonic hedgehog signaling, and altered cell-cycle progression. Furthermore, the ALS-linked variants induced intracellular calcium overload followed by Aurora kinase A (AurA)-histone deacetylase (HDAC)6 activation, resulting in ciliary disassembly. These defects were restored by treatment with the intracellular Ca2+ chelator, BAPTA. We also found that NEK1 variants cause decreased α-tubulin acetylation, mitochondrial alteration, and impaired DNA damage response (DDR). Notably, drug treatment to inhibit HDAC6 restored the NEK1-dependent deficits in patient fibroblasts. And, we confirmed that data found in patient fibroblasts were reproduced in iPSC-MNs model.
Conclusions: Our results suggest that NEK1 contributes to ALS pathogenesis through the LOF mechanism, and HDAC6 inhibition provides an attractive therapeutic strategy for NEK1 variants associated ALS treatment.
Keywords: NEK1; Amyotrophic lateral sclerosis; Cell cycle; DNA damage response; Microtubule; Mitochondria; Primary cilia.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: All participants provided written informed consent for the use of human-derived materials and clinical and genetic information for research purposes. This study was reviewed and approved by the IRB of Hanyang University. Consent for publication: The authors consent to the publication of this manuscript. Confidential patient data were not included in this study. Competing interests: The authors declare that they have no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
