

The gene regulatory mechanisms shaping the heterogeneity of venom production in the Cape coral snake
- PMID: 40390047
- PMCID: PMC12087220
- DOI: 10.1186/s13059-025-03602-w
The gene regulatory mechanisms shaping the heterogeneity of venom production in the Cape coral snake
Abstract
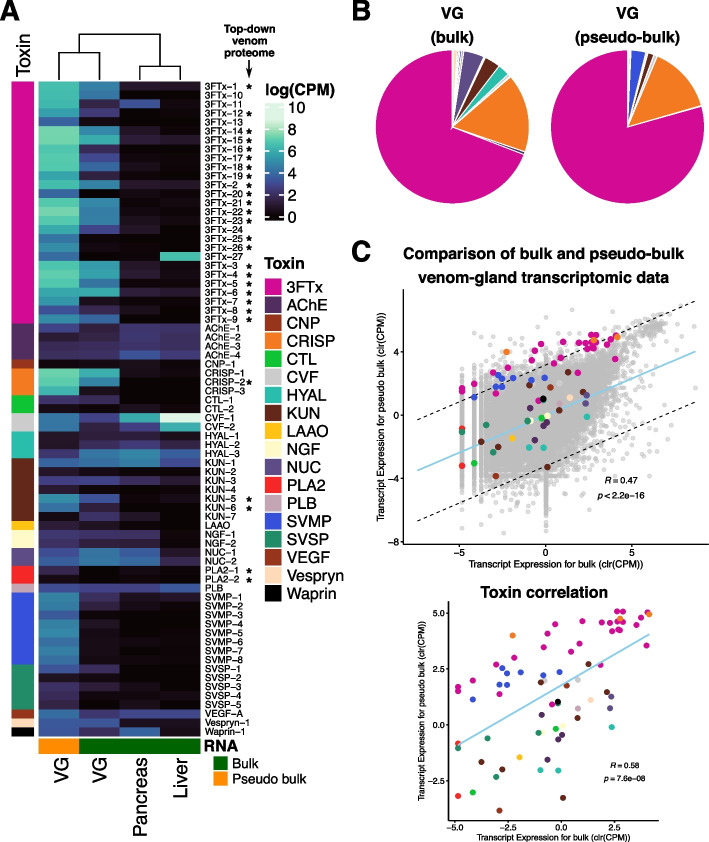
Background: Venoms and their associated glands and delivery structures have evolved numerous times among animals. Within these venom systems, the molecular, cellular, and morphological components interact and co-evolve to generate distinct, venom phenotypes that are increasingly recognized as models for studying adaptive evolution. However, toxins are often unevenly distributed across venom-producing tissues in patterns that are not necessarily adaptive but instead likely result from constraints associated with protein secretion.
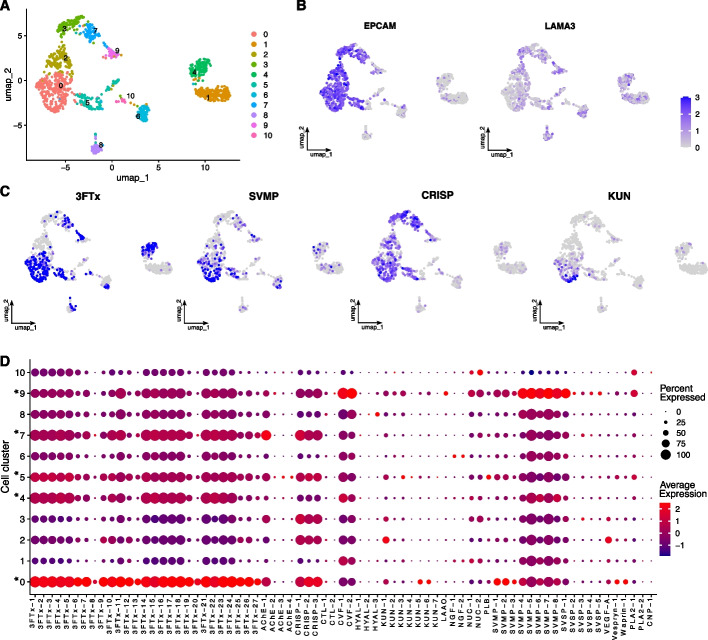
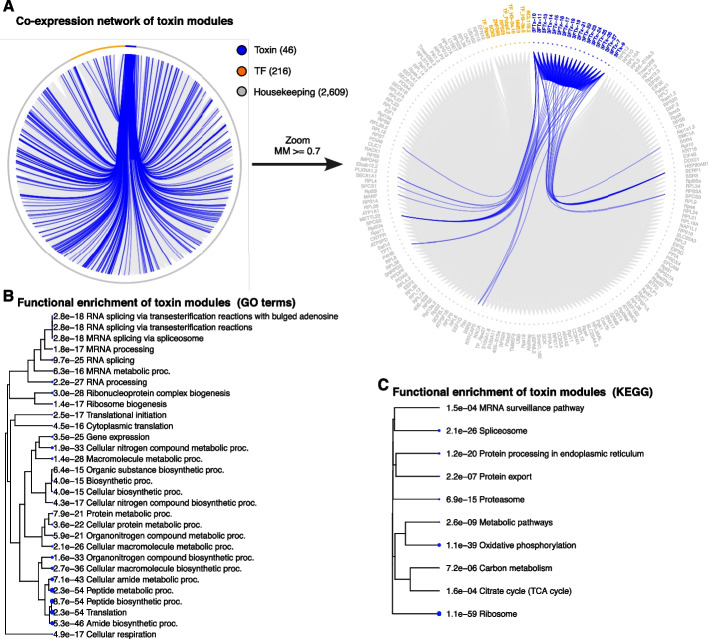
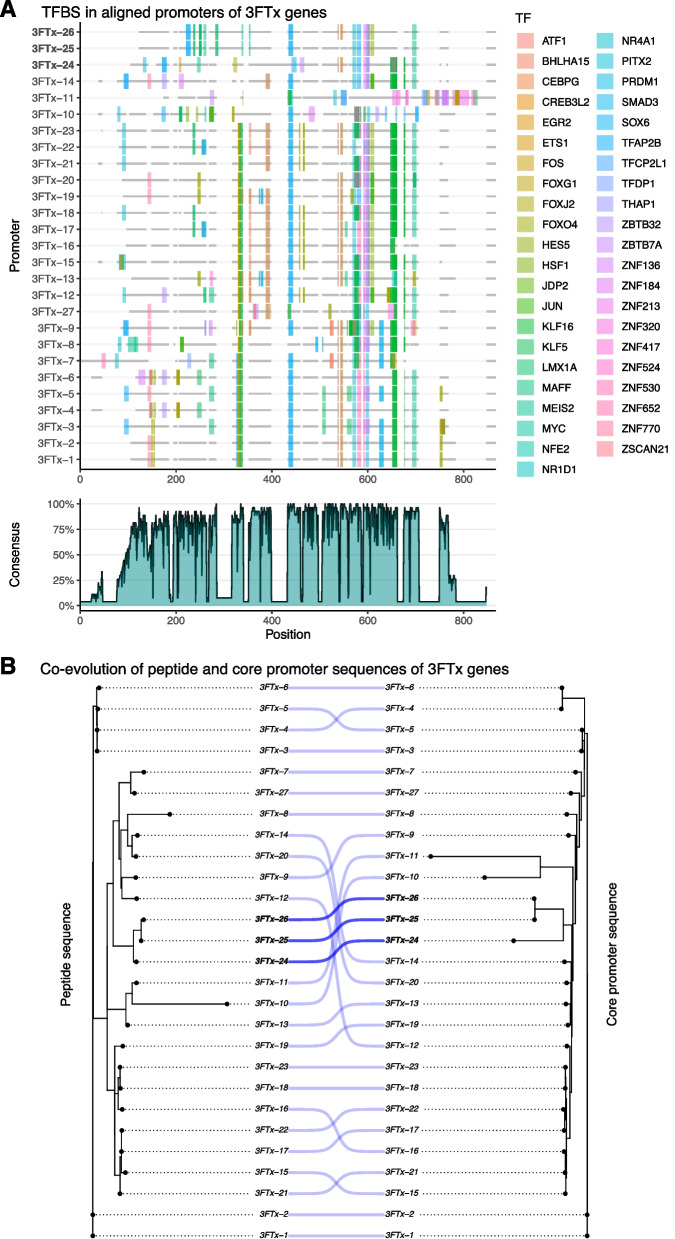
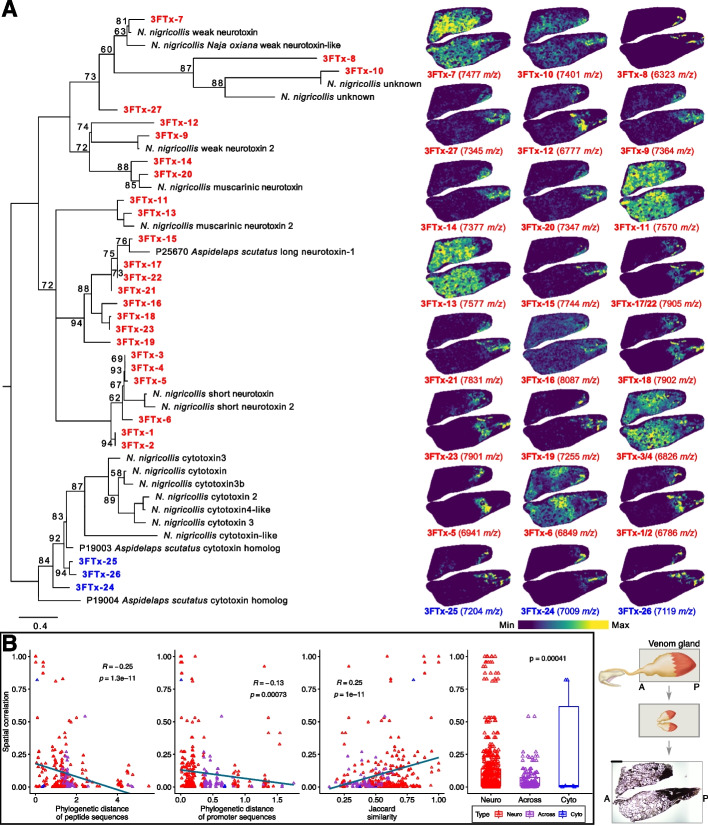
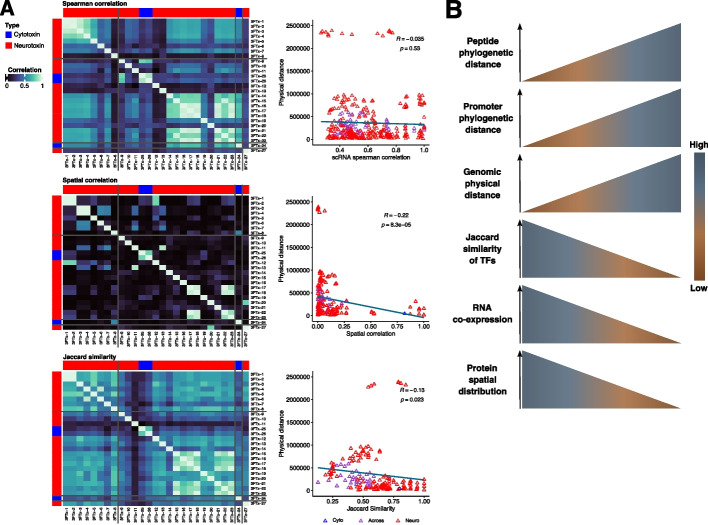
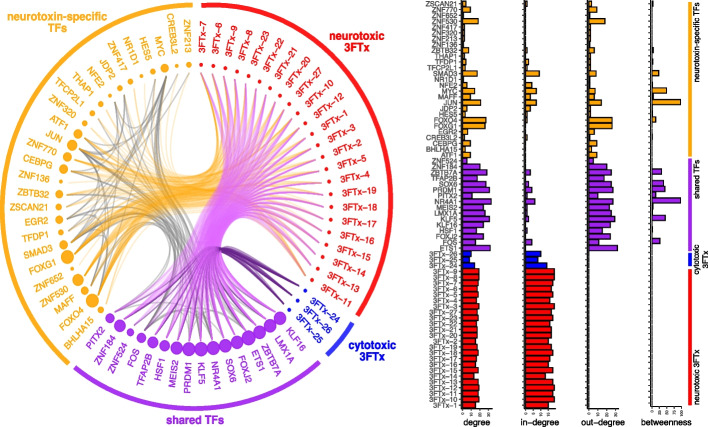
Results: We generate a high-quality draft genome of the Cape coral snake (Aspidelaps lubricus) and combine analyses of venom gland single-cell RNA-seq data with spatial venom gland in situ toxin distributions. Our results reveal that while different toxin families are produced by distinct populations of cells, toxin expression is fine-tuned by regulatory modules that result in further specialization of toxin production within each cell population. We also find that the evolution of regulatory elements closely mirrors the evolution of their associated toxin genes, resulting in spatial association of closely related and functionally similar toxins in the venom gland. While this compartmentalization is non-adaptive, the modularity of the underlying regulatory network likely facilitated the repeated evolution of defensive venom in spitting cobras.
Conclusions: Our results provide new insight into the variability of toxin regulation across snakes, reveal the molecular mechanisms underlying the heterogeneous toxin production in snake venom glands, and provide an example of how constraints can result in non-adaptive character states that appear to be adaptive, which may nevertheless facilitate evolutionary innovation and novelty.
Keywords: Elapidae; Gene regulatory network; Genomics; Mass spectrometry imaging; Toxin; Venomics.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Competing interests: The authors declare no competing interests.
Figures

Similar articles
-
Distinct regulatory networks control toxin gene expression in elapid and viperid snakes.BMC Genomics. 2024 Feb 16;25(1):186. doi: 10.1186/s12864-024-10090-y. BMC Genomics. 2024. PMID: 38365592 Free PMC article.
-
New insights into the phylogeographic distribution of the 3FTx/PLA2 venom dichotomy across genus Micrurus in South America.J Proteomics. 2019 May 30;200:90-101. doi: 10.1016/j.jprot.2019.03.014. Epub 2019 Apr 1. J Proteomics. 2019. PMID: 30946991
-
The venom-gland transcriptome of the eastern coral snake (Micrurus fulvius) reveals high venom complexity in the intragenomic evolution of venoms.BMC Genomics. 2013 Aug 2;14:531. doi: 10.1186/1471-2164-14-531. BMC Genomics. 2013. PMID: 23915248 Free PMC article.
-
Venomous snakes of Costa Rica: biological and medical implications of their venom proteomic profiles analyzed through the strategy of snake venomics.J Proteomics. 2014 Jun 13;105:323-39. doi: 10.1016/j.jprot.2014.02.020. Epub 2014 Feb 24. J Proteomics. 2014. PMID: 24576642 Review.
-
Venoms of Micrurus coral snakes: Evolutionary trends in compositional patterns emerging from proteomic analyses.Toxicon. 2016 Nov;122:7-25. doi: 10.1016/j.toxicon.2016.09.008. Epub 2016 Sep 15. Toxicon. 2016. PMID: 27641749 Review.
References
-
- Asp M, Giacomello S, Larsson L, Wu C, Fürth D, Qian X, et al. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell. 2019;179(7):1647–60. - PubMed
-
- Jiang S, Huang Z, Li Y, Yu C, Yu H, Ke Y, et al. Single-cell chromatin accessibility and transcriptome atlas of mouse embryos. Cell Rep. 2023;42(3):112210. - PubMed
-
- Snijder B, Pelkmans L. Origins of regulated cell-to-cell variability. Nat Rev Mol Cell Biol. 2011;12(2):119–25. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
