

Lactate dehydrogenase A-coupled NAD+ regeneration is critical for acute myeloid leukemia cell survival
- PMID: 40390151
- PMCID: PMC12090514
- DOI: 10.1186/s40170-025-00392-4
Lactate dehydrogenase A-coupled NAD+ regeneration is critical for acute myeloid leukemia cell survival
Abstract
Background: Enhanced glycolysis plays a pivotal role in fueling the aberrant proliferation, survival and therapy resistance of acute myeloid leukemia (AML) cells. Here, we aimed to elucidate the extent of glycolysis dependence in AML by focusing on the role of lactate dehydrogenase A (LDHA), a key glycolytic enzyme converting pyruvate to lactate coupled with the recycling of NAD+.
Methods: We compared the glycolytic activity of primary AML patient samples to protein levels of metabolic enzymes involved in central carbon metabolism including glycolysis, glutaminolysis and the tricarboxylic acid cycle. To evaluate the therapeutic potential of targeting glycolysis in AML, we treated AML primary patient samples and cell lines with pharmacological inhibitors of LDHA and monitored cell viability. Glycolytic activity and mitochondrial oxygen consumption were analyzed in AML patient samples and cell lines post-LDHA inhibition. Perturbations in global metabolite levels and redox balance upon LDHA inhibition in AML cells were determined by mass spectrometry, and ROS levels were measured by flow cytometry.
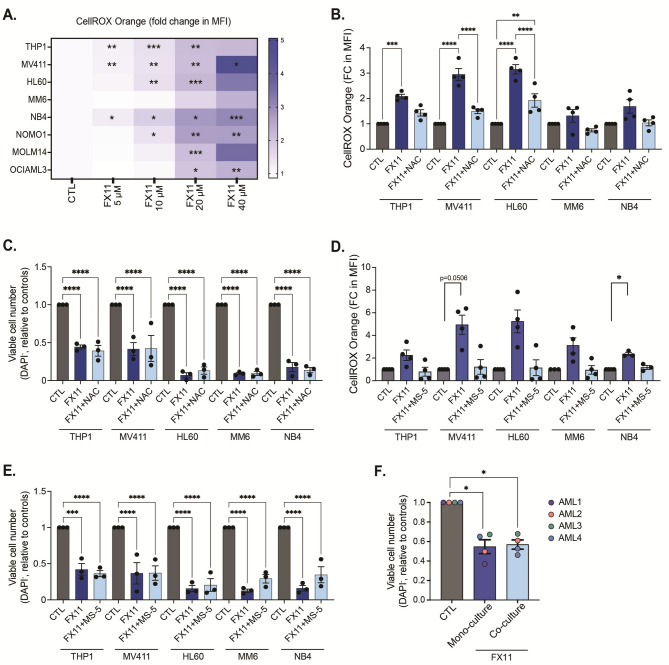
Results: Among metabolic enzymes, we found that LDHA protein levels had the strongest positive correlation with glycolysis in AML patient cells. Blocking LDHA activity resulted in a strong growth inhibition and cell death induction in AML cell lines and primary patient samples, while healthy hematopoietic stem and progenitor cells remained unaffected. Investigation of the underlying mechanisms showed that LDHA inhibition reduces glycolytic activity, lowers levels of glycolytic intermediates, decreases the cellular NAD+ pool, boosts OXPHOS activity and increases ROS levels. This increase in ROS levels was however not linked to the observed AML cell death. Instead, we found that LDHA is essential to maintain a correct NAD+/NADH ratio in AML cells. Continuous intracellular NAD+ supplementation via overexpression of water-forming NADH oxidase from Lactobacillus brevis in AML cells effectively increased viable cell counts and prevented cell death upon LDHA inhibition.
Conclusions: Collectively, our results demonstrate that AML cells critically depend on LDHA to maintain an adequate NAD+/NADH balance in support of their abnormal glycolytic activity and biosynthetic demands, which cannot be compensated for by other cellular NAD+ recycling systems. These findings also highlight LDHA inhibition as a promising metabolic strategy to eradicate leukemic cells.
Keywords: Acute myeloid leukemia; Cancer metabolism; Glycolysis; Lactate dehydrogenase A; NAD+; Redox balance.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures




Update of
-
Lactate dehydrogenase A-coupled NAD+ regeneration is critical for acute myeloid leukemia cell survival.bioRxiv [Preprint]. 2025 Mar 7:2025.03.03.641255. doi: 10.1101/2025.03.03.641255. bioRxiv. 2025. Update in: Cancer Metab. 2025 May 19;13(1):22. doi: 10.1186/s40170-025-00392-4. PMID: 40093073 Free PMC article. Updated. Preprint.
Similar articles
-
Lactate dehydrogenase A-coupled NAD+ regeneration is critical for acute myeloid leukemia cell survival.bioRxiv [Preprint]. 2025 Mar 7:2025.03.03.641255. doi: 10.1101/2025.03.03.641255. bioRxiv. 2025. Update in: Cancer Metab. 2025 May 19;13(1):22. doi: 10.1186/s40170-025-00392-4. PMID: 40093073 Free PMC article. Updated. Preprint.
-
Amino acid transporter LAT1 (SLC7A5) promotes metabolic rewiring in TNBC progression through the L-Trp/QPRT/NAD+ pathway.J Exp Clin Cancer Res. 2025 Jul 3;44(1):190. doi: 10.1186/s13046-025-03446-z. J Exp Clin Cancer Res. 2025. PMID: 40611146 Free PMC article.
-
Downregulation of LATS1/2 Drives Endothelial Senescence-Associated Stemness (SAS) and Atherothrombotic Lesion Formation.bioRxiv [Preprint]. 2025 Jun 21:2025.06.19.660635. doi: 10.1101/2025.06.19.660635. bioRxiv. 2025. PMID: 40667385 Free PMC article. Preprint.
-
Impact of residual disease as a prognostic factor for survival in women with advanced epithelial ovarian cancer after primary surgery.Cochrane Database Syst Rev. 2022 Sep 26;9(9):CD015048. doi: 10.1002/14651858.CD015048.pub2. Cochrane Database Syst Rev. 2022. PMID: 36161421 Free PMC article.
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
Cited by
-
Mitochondrial metabolism and cancer therapeutic innovation.Signal Transduct Target Ther. 2025 Aug 4;10(1):245. doi: 10.1038/s41392-025-02311-x. Signal Transduct Target Ther. 2025. PMID: 40754534 Free PMC article. Review.
-
Living on the Edge: ROS Homeostasis in Cancer Cells and Its Potential as a Therapeutic Target.Antioxidants (Basel). 2025 Aug 16;14(8):1002. doi: 10.3390/antiox14081002. Antioxidants (Basel). 2025. PMID: 40867898 Free PMC article. Review.
References
-
- Warburg O. Über Den Stoffwechsel der carcinomzelle. Naturwissenschaften. 1924;12:1131–7.
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
