

Whole genome sequencing improves tissue-of-origin diagnosis and treatment options for cancer of unknown primary
- PMID: 40393956
- PMCID: PMC12092688
- DOI: 10.1038/s41467-025-59661-x
Whole genome sequencing improves tissue-of-origin diagnosis and treatment options for cancer of unknown primary
Abstract
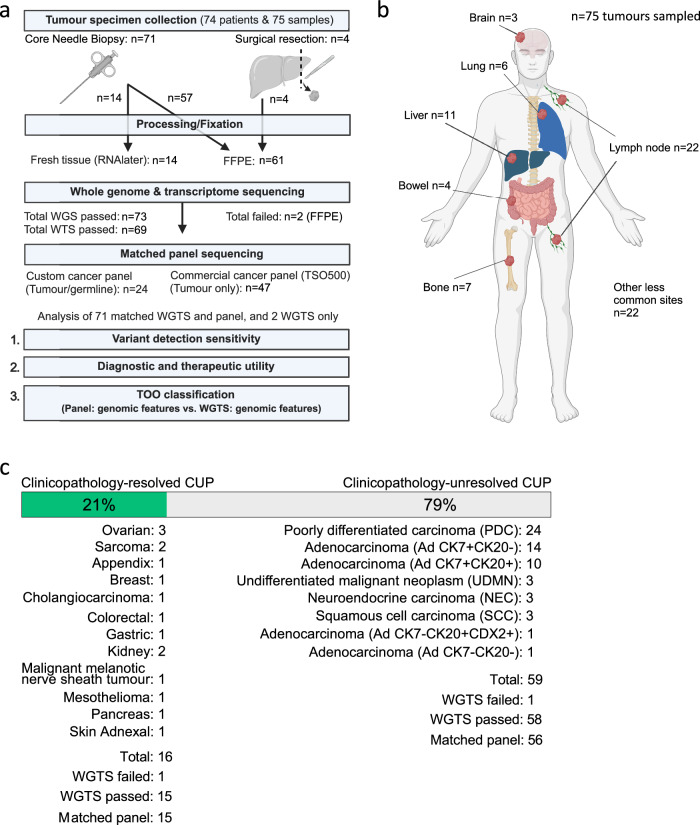
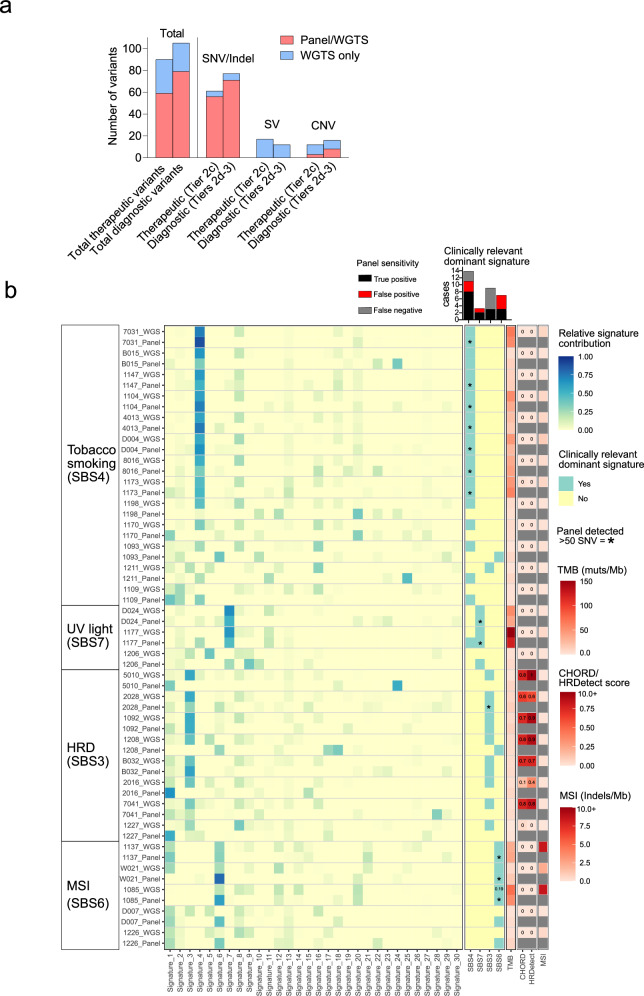
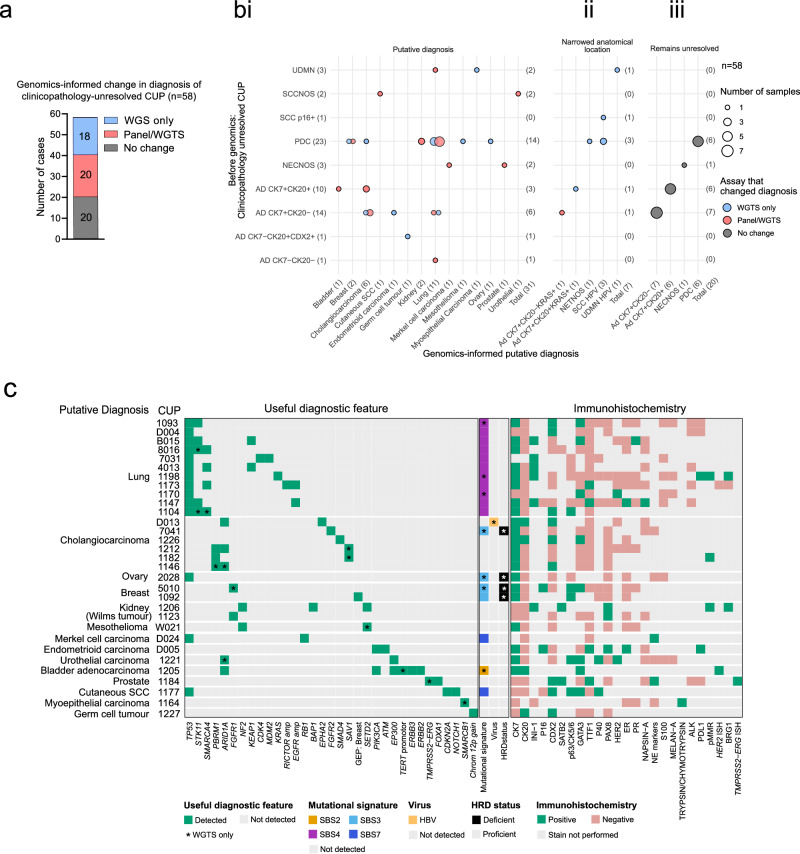
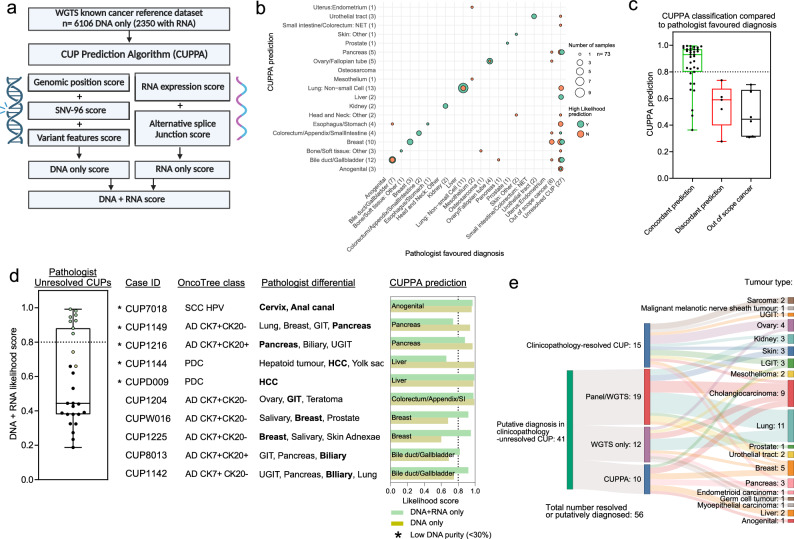
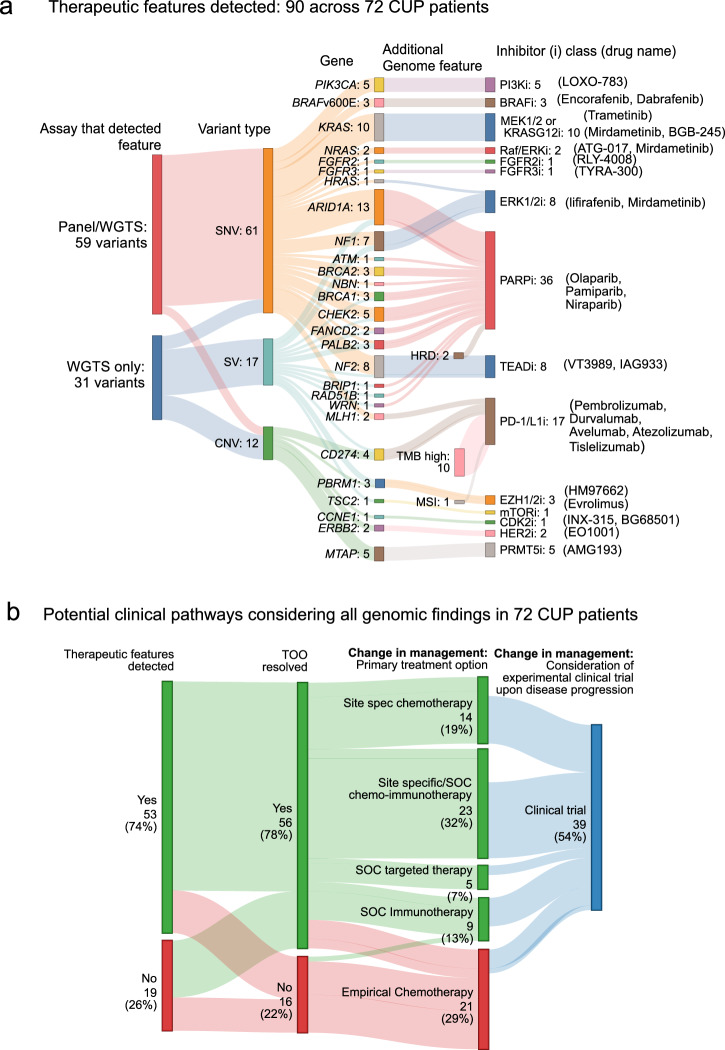
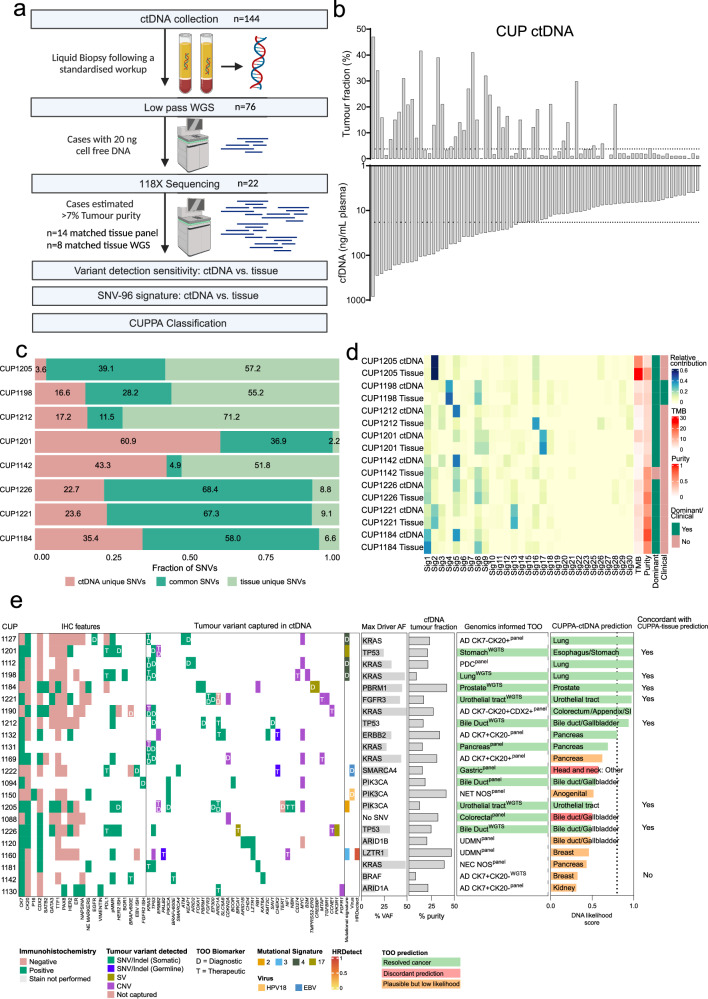
Genomics can inform both tissue-of-origin (TOO) and precision treatments for patients with cancer of unknown primary (CUP). Here, we use whole genome and transcriptome sequencing (WGTS) for 72 patients and show diagnostic superiority of WGTS over panel testing (386-523 genes) in 71 paired cases. WGTS detects all reportable DNA features found by panel as well as additional mutations of diagnostic or therapeutic relevance in 76% of cases. Curated WGTS features and a CUP prediction algorithm (CUPPA) trained on WGTS data of known cancer types informs TOO in 71% of cases otherwise undiagnosed by clinicopathology review. WGTS informs treatments for 79% of patients, compared to 59% by panel testing. Finally, WGS of cell-free DNA (cfDNA) from patients with a high cfDNA tumour fraction (>7%), enables high-likelihood CUPPA predictions in 41% of cases. WGTS is therefore superior to panel testing, broadens treatment options, and is feasible using routine pathology samples and cfDNA.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: All authors declare no competing interests.
Figures
References
-
- Sivakumaran, T., Tothill, R. W. & Mileshkin, L. R. The evolution of molecular management of carcinoma of unknown primary. Curr. Opin. Oncol.36, 456–464 (2024). - PubMed
-
- Kramer, A., et al. Molecularly guided therapy versus chemotherapy after disease control in unfavourable cancer of unknown primary (CUPISCO): an open-label, randomised, phase 2 study. Lancet404, 527–539 (2024). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
