

Human mitochondrial ferritin exhibits highly unusual iron-O2 chemistry distinct from that of cytosolic ferritins
- PMID: 40393986
- PMCID: PMC12092714
- DOI: 10.1038/s41467-025-59463-1
Human mitochondrial ferritin exhibits highly unusual iron-O2 chemistry distinct from that of cytosolic ferritins
Abstract
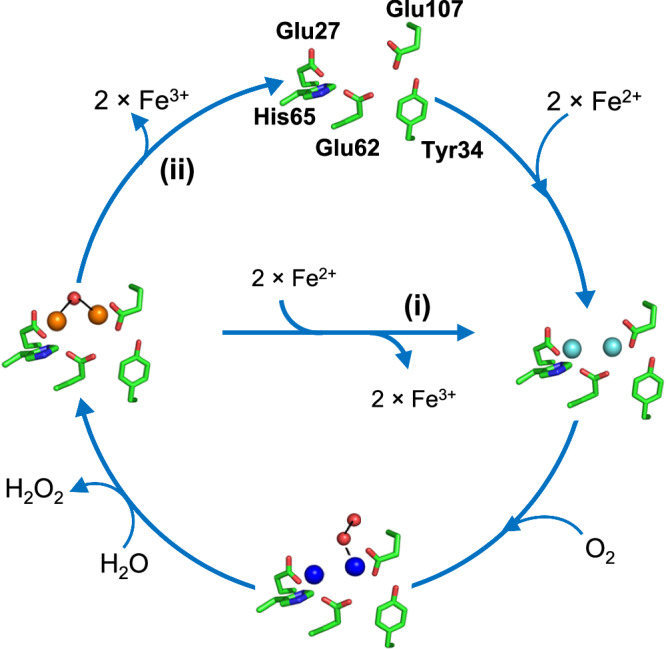
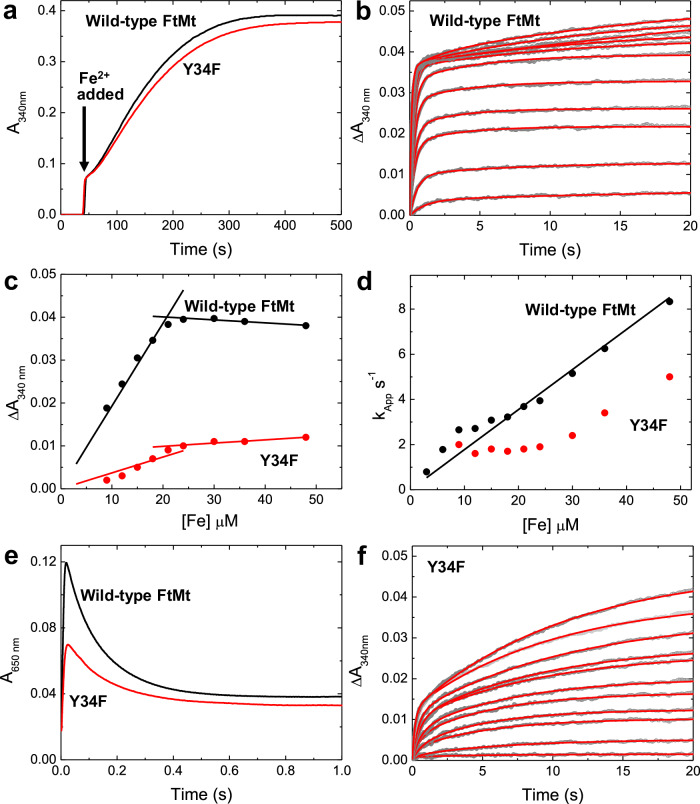
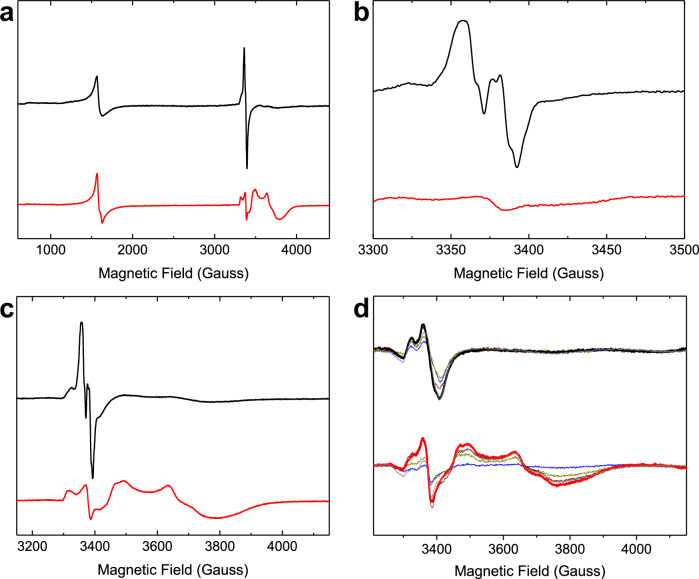
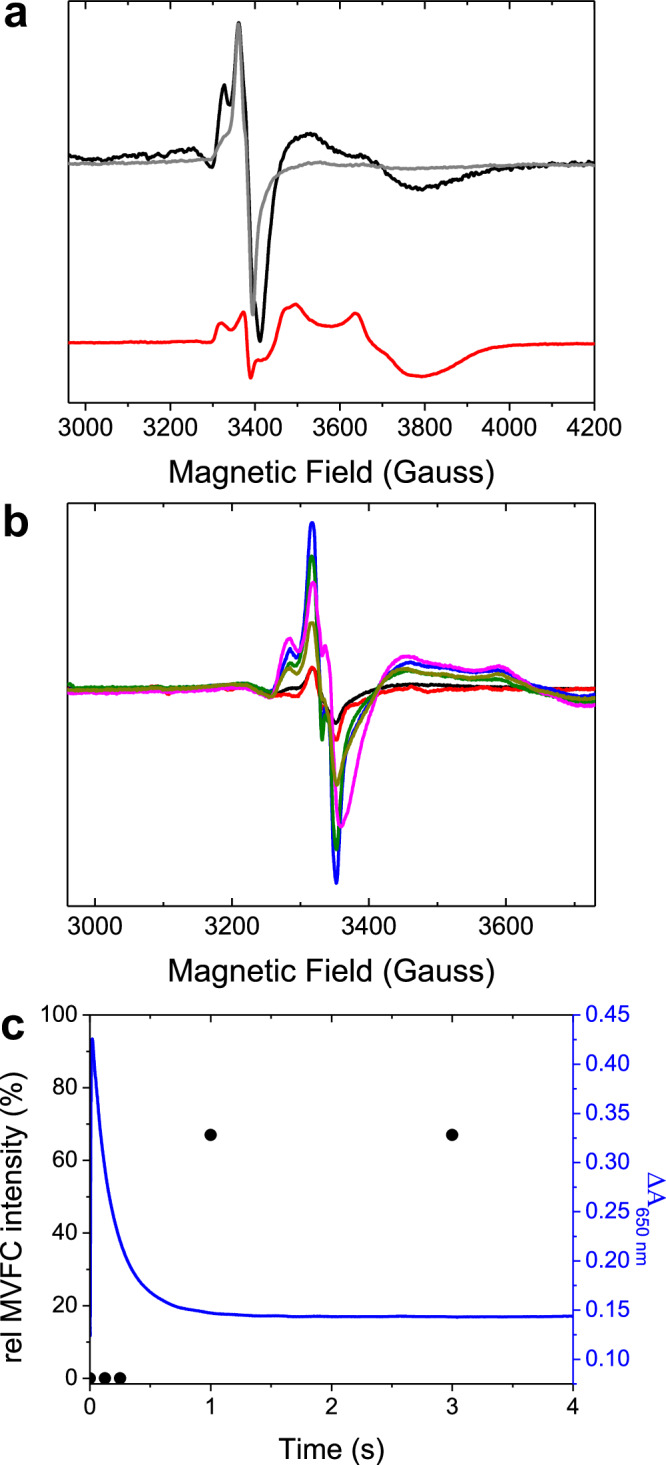
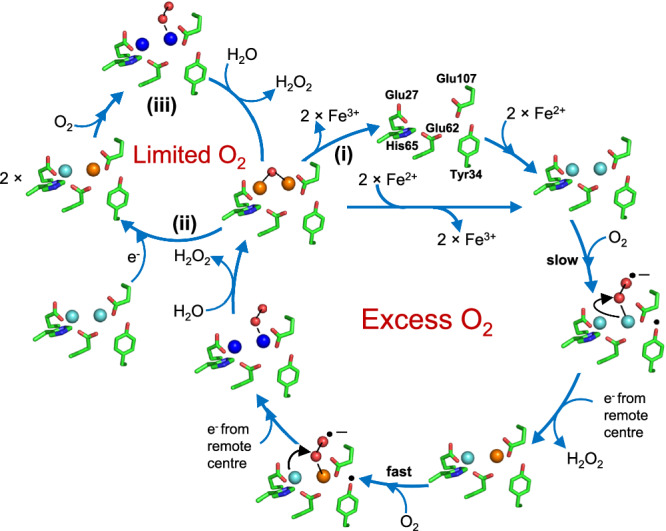
Ferritins are ubiquitous proteins that function in iron storage/detoxification by catalyzing the oxidation of Fe2+ ions and solubilizing the resulting Fe3+-oxo mineral. Mammalian tissues that are metabolically highly active contain, in addition to the widespread cytosolic ferritin, a ferritin that is localized to mitochondria. Mitochondrial ferritin (FtMt) protects against oxidative stress and is found at higher levels in diseases associated with abnormal iron accumulation, including Alzheimer's and Parkinson's. Here we demonstrate that, despite 80% sequence identity with cytosolic human H-chain ferritin, Fe2+ oxidation at the catalytic diiron ferroxidase center of FtMt proceeds via a distinct mechanism. This involves a mixed-valent ferroxidase center (MVFC) that is readily detected under the O2-limiting conditions typical of mitochondria, and formation of a radical on a strictly conserved Tyr residue (Tyr34) that is key for the activation of O2 and stability of the MVFC. The possible origin of the mechanistic differences exhibited by the highly-related human mitochondrial and cytosolic H-chain ferritins is explored.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Unique iron binding and oxidation properties of human mitochondrial ferritin: a comparative analysis with Human H-chain ferritin.J Mol Biol. 2005 Apr 1;347(3):543-54. doi: 10.1016/j.jmb.2005.01.007. J Mol Biol. 2005. PMID: 15755449
-
Iron Oxidation and Core Formation in Recombinant Heteropolymeric Human Ferritins.Biochemistry. 2017 Aug 1;56(30):3900-3912. doi: 10.1021/acs.biochem.7b00024. Epub 2017 Jul 18. Biochemistry. 2017. PMID: 28636371 Free PMC article.
-
Reaction of O2 with a diiron protein generates a mixed-valent Fe2+/Fe3+ center and peroxide.Proc Natl Acad Sci U S A. 2019 Feb 5;116(6):2058-2067. doi: 10.1073/pnas.1809913116. Epub 2019 Jan 18. Proc Natl Acad Sci U S A. 2019. PMID: 30659147 Free PMC article.
-
Iron mineralization and core dissociation in mammalian homopolymeric H-ferritin: Current understanding and future perspectives.Biochim Biophys Acta Gen Subj. 2020 Nov;1864(11):129700. doi: 10.1016/j.bbagen.2020.129700. Epub 2020 Aug 14. Biochim Biophys Acta Gen Subj. 2020. PMID: 32798636 Review.
-
Mechanisms of iron mineralization in ferritins: one size does not fit all.J Biol Inorg Chem. 2014 Aug;19(6):775-85. doi: 10.1007/s00775-014-1136-3. Epub 2014 Apr 19. J Biol Inorg Chem. 2014. PMID: 24748222 Review.
References
-
- Beinert, H., Holm, R. H. & Munck, E. Iron-sulfur clusters: Nature’s modular, multipurpose structures. Science277, 653–659 (1997). - PubMed
-
- Gamba, I., Codola, Z., Lloret-Fillol, J. & Costas, M. Making and breaking of the O-O bond at iron complexes. Coord. Chem. Rev.334, 2–24 (2017).
-
- Gayer, K. H. & Wootner, L. The hydrolysis of ferrous chloride at 25 degrees. J. Am. Chem. Soc.78, 3944–3946 (1956).
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
