

Empagliflozin enhances metabolic efficiency and improves left ventricular hypertrophy in a hypertrophic cardiomyopathy mouse model
- PMID: 40396194
- PMCID: PMC12539926
- DOI: 10.1093/eurheartj/ehaf324
Empagliflozin enhances metabolic efficiency and improves left ventricular hypertrophy in a hypertrophic cardiomyopathy mouse model
Abstract
Background and aims: Hypertrophic cardiomyopathy (HCM) is a genetic cardiac disorder characterized by left ventricular hypertrophy (LVH), diastolic dysfunction, and impaired metabolic efficiency. This study investigates the therapeutic potential of the sodium-glucose cotransporter 2 inhibitor (SGLT2i) empagliflozin (EMPA) in ameliorating these pathological features in a mouse model carrying the myosin R403Q mutation.
Methods: Male mice harbouring the R403Q mutation were treated with EMPA for 16 weeks. Multi-nuclear magnetic resonance spectroscopy (31P, 13C, and 23Na MRS), echocardiography, transcriptomic, proteomic, and phosphoproteomic profiling were utilized to assess metabolic, structural, and functional changes.
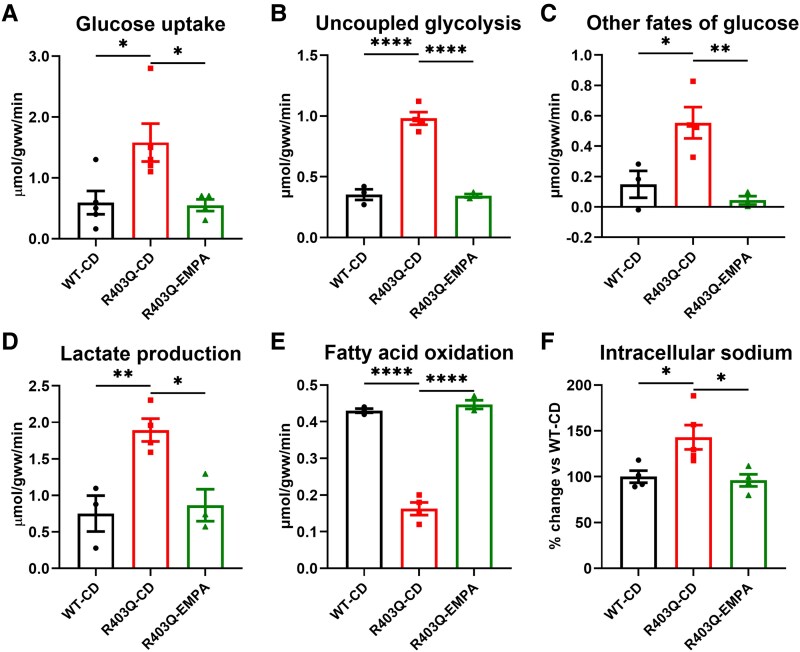
Results: Empagliflozin facilitated the coupling of glycolysis with glucose oxidation and normalized elevated intracellular sodium levels. Treatment resulted in a significant reduction in LVH and myocardial fibrosis as evidenced by echocardiography and histopathology. These structural improvements correlated with enhancements in mitochondrial adenosine triphosphate (ATP) synthesis, fatty acid oxidation, and branched-chain amino acid catabolism. Furthermore, EMPA improved left ventricular diastolic function and contractile reserve, underscored by improved ATP production and reduced energy cost of contraction. Notably, these benefits were linked to down-regulation of the mammalian target of rapamycin signalling pathway and normalization of myocardial substrate metabolic fluxes.
Conclusions: Empagliflozin significantly mitigates structural and metabolic dysfunctions in a mouse model of HCM, underscoring its potential as a therapeutic agent for managing this condition. These findings suggest broader applicability of SGLT2i in cardiovascular diseases, including those due to myocardial-specific mutations, warranting further clinical investigation.
Keywords: Branched-chain amino acids; Cardiac energetics; Cardiac metabolism; Hypertrophic cardiomyopathy; Metabolic reprogramming; SGLT2 inhibition; Uncoupled glycolysis; mTOR.
© The Author(s) 2025. Published by Oxford University Press on behalf of the European Society of Cardiology.
Figures






References
-
- Toepfer CN, Garfinkel AC, Venturini G, Wakimoto H, Repetti G, Alamo L, et al. Myosin sequestration regulates sarcomere function, cardiomyocyte energetics, and metabolism, informing the pathogenesis of hypertrophic cardiomyopathy. Circulation 2020;141:828–42. 10.1161/CIRCULATIONAHA.119.042339 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
