

Clonal tracing with somatic epimutations reveals dynamics of blood ageing
- PMID: 40399669
- PMCID: PMC12240852
- DOI: 10.1038/s41586-025-09041-8
Clonal tracing with somatic epimutations reveals dynamics of blood ageing
Abstract
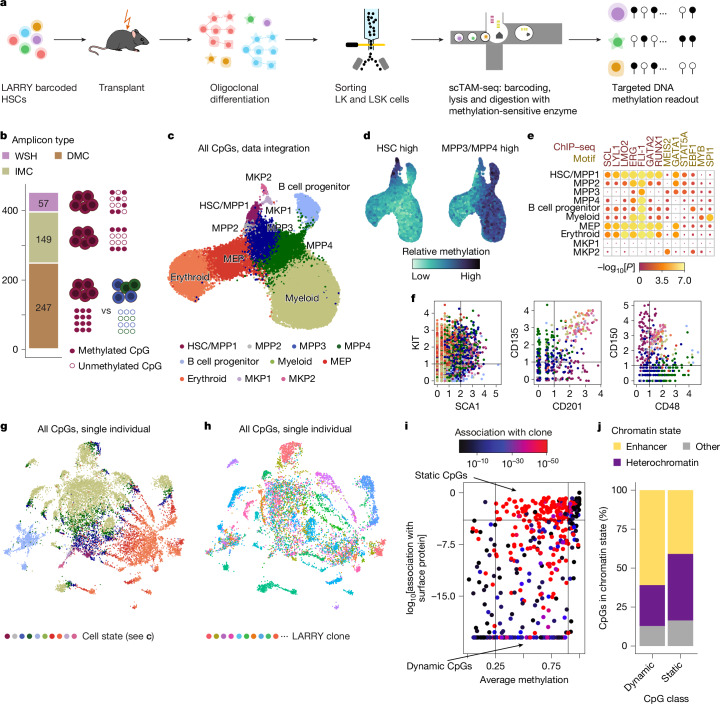
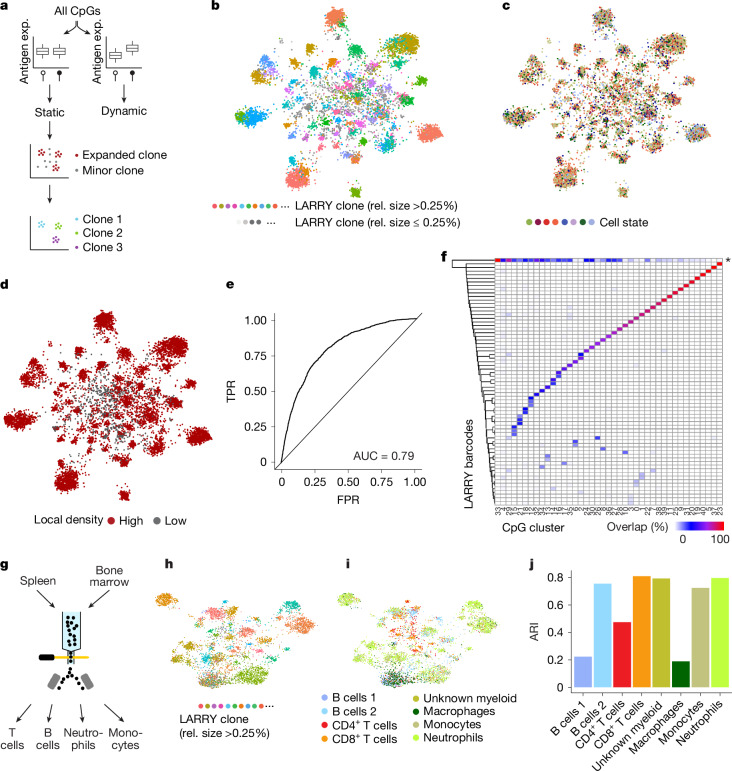
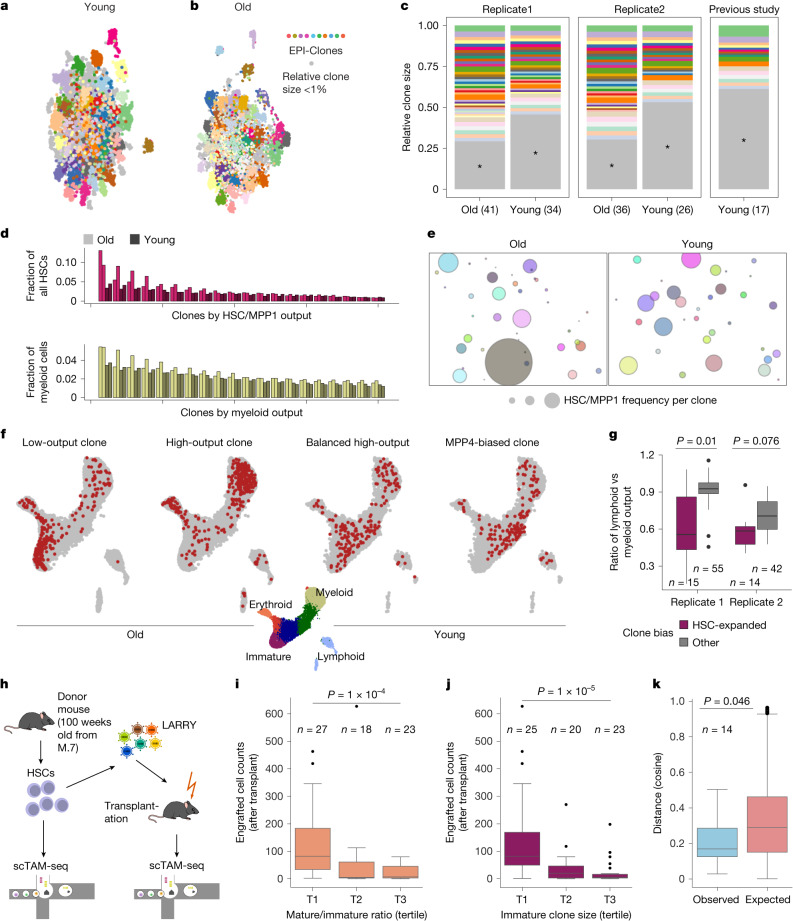
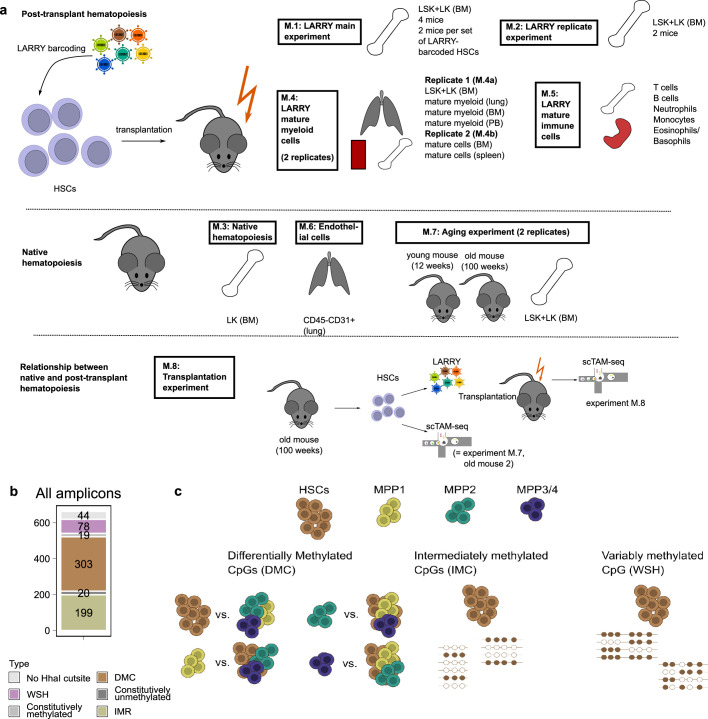
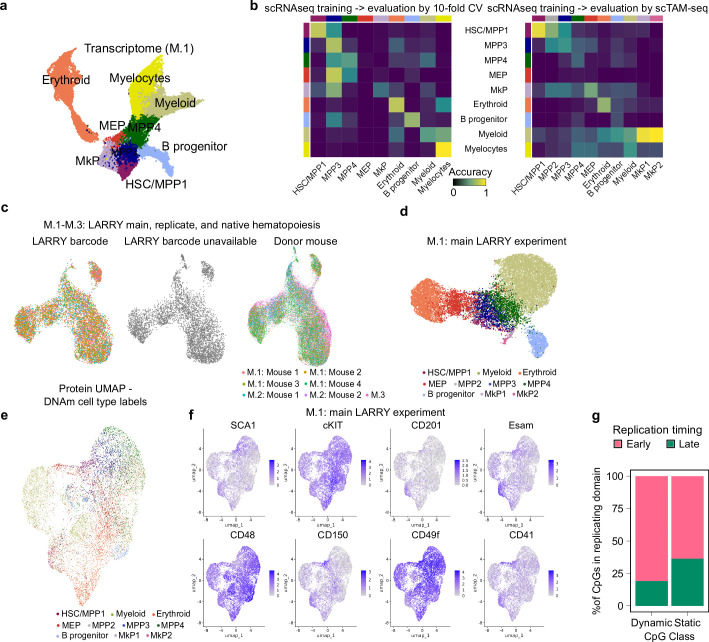
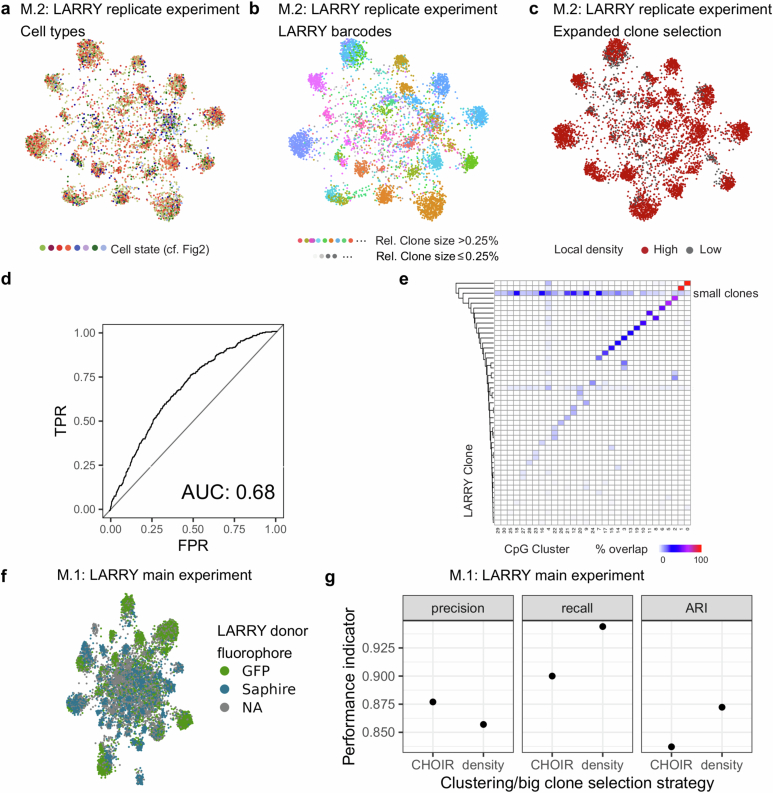
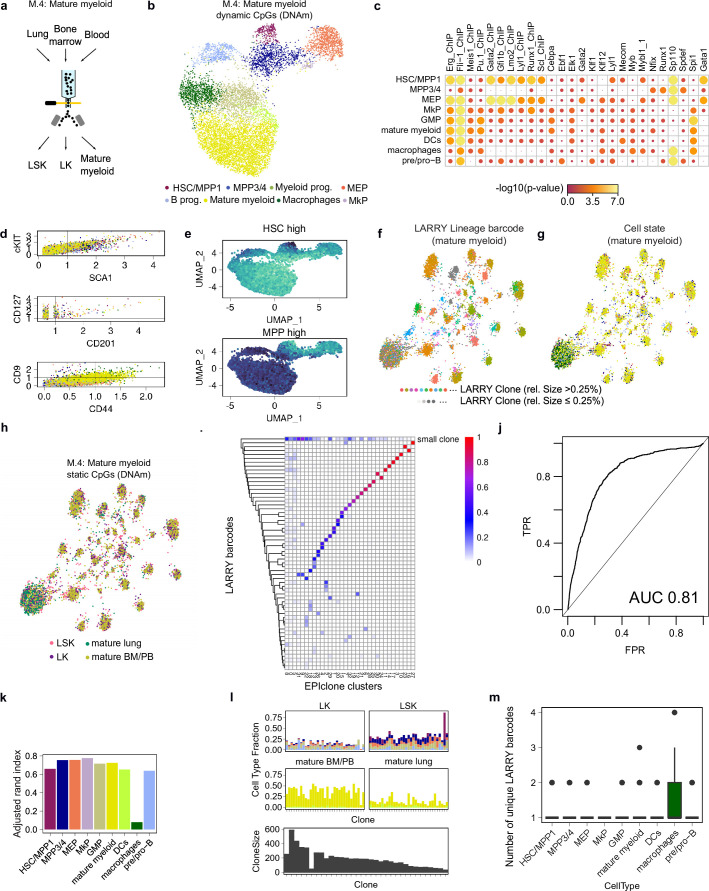
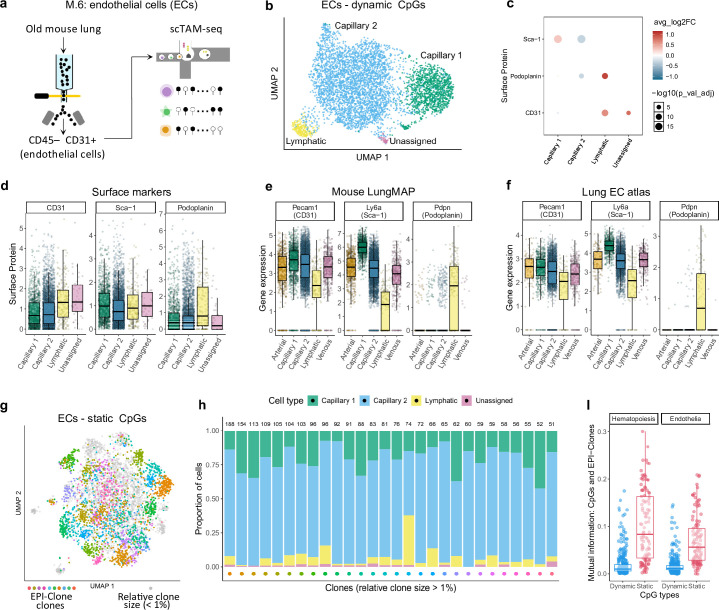
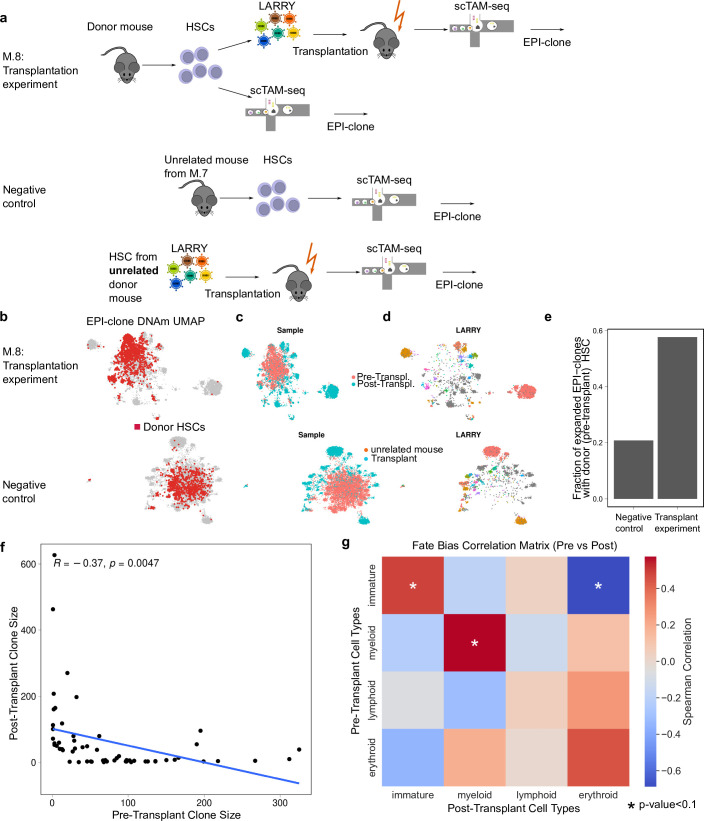
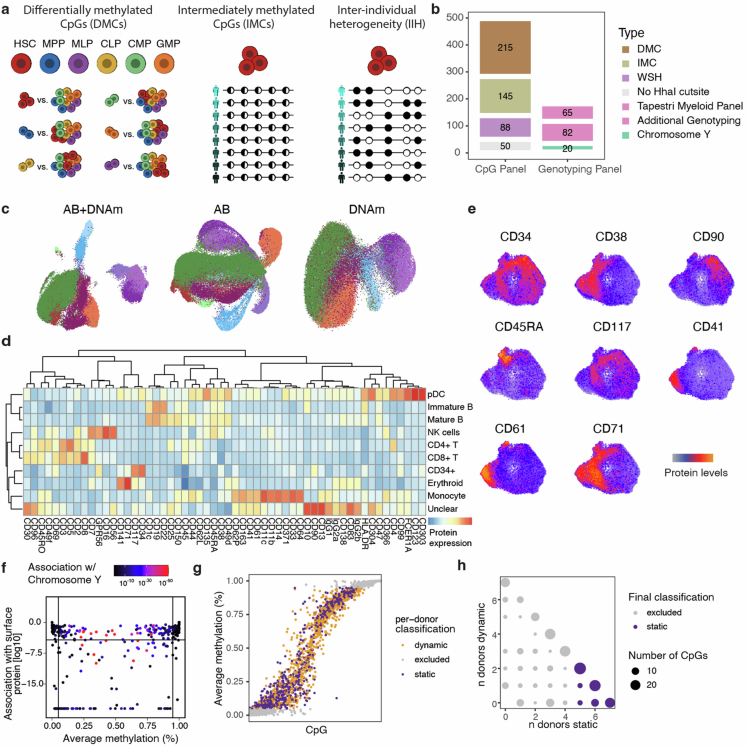
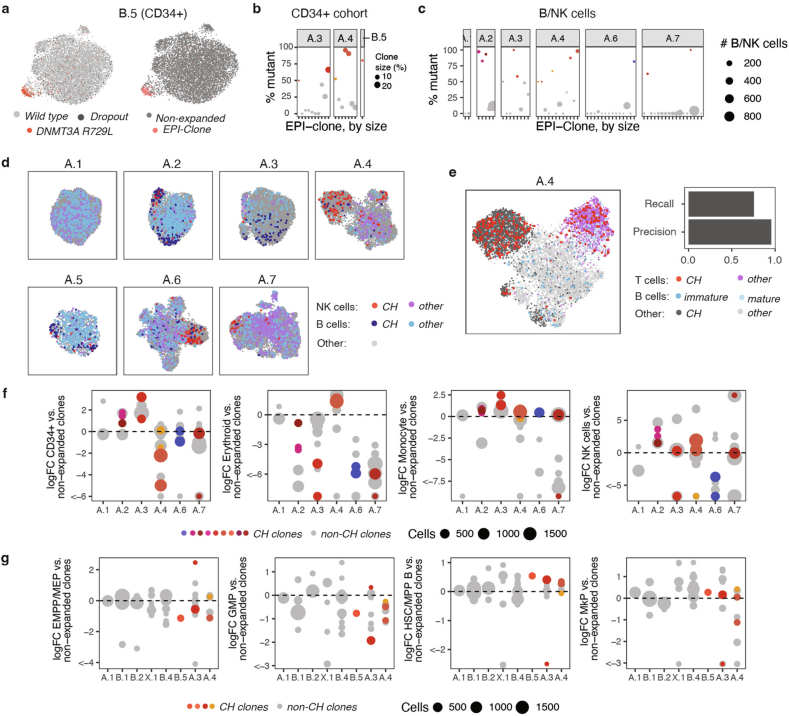
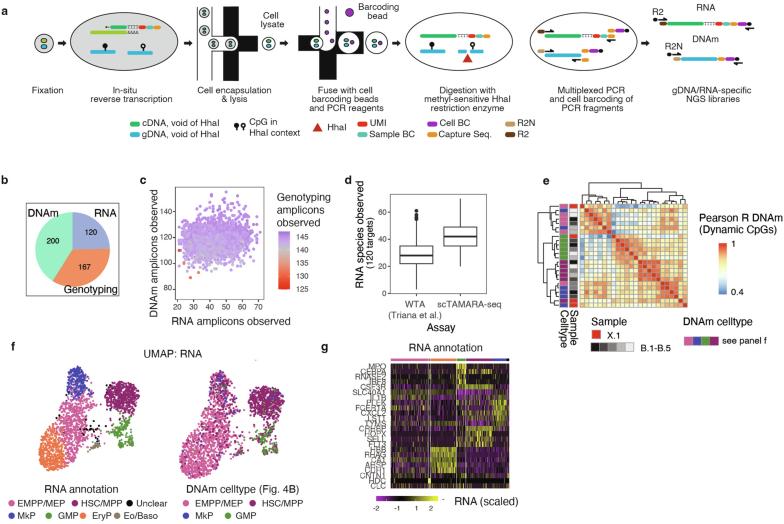
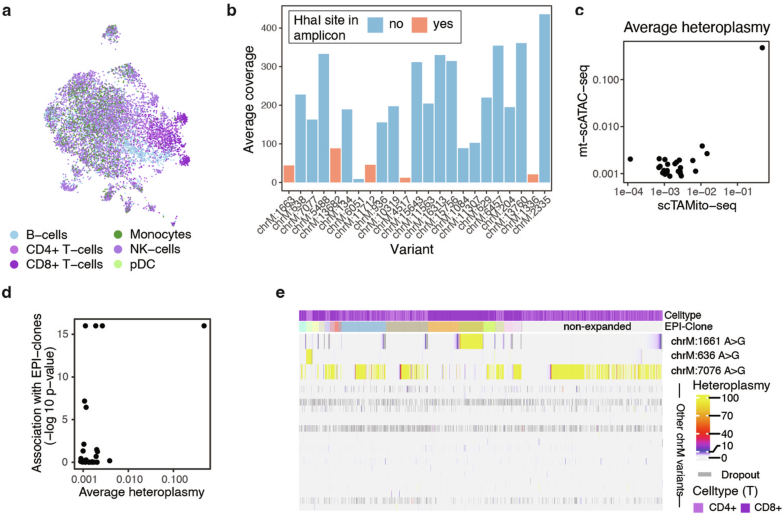
Current approaches used to track stem cell clones through differentiation require genetic engineering1,2 or rely on sparse somatic DNA variants3,4, which limits their wide application. Here we discover that DNA methylation of a subset of CpG sites reflects cellular differentiation, whereas another subset undergoes stochastic epimutations and can serve as digital barcodes of clonal identity. We demonstrate that targeted single-cell profiling of DNA methylation5 at single-CpG resolution can accurately extract both layers of information. To that end, we develop EPI-Clone, a method for transgene-free lineage tracing at scale. Applied to mouse and human haematopoiesis, we capture hundreds of clonal differentiation trajectories across tens of individuals and 230,358 single cells. In mouse ageing, we demonstrate that myeloid bias and low output of old haematopoietic stem cells6 are restricted to a small number of expanded clones, whereas many functionally young-like clones persist in old age. In human ageing, clones with and without known driver mutations of clonal haematopoieis7 are part of a spectrum of age-related clonal expansions that display similar lineage biases. EPI-Clone enables accurate and transgene-free single-cell lineage tracing on hematopoietic cell state landscapes at scale.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: A.R.-F. serves as an advisor for Retro Bio. Parts of this study have been supported with reagents donated by Mission Bio. The other authors declare no competing interests.
Figures


Update of
-
Somatic epimutations enable single-cell lineage tracing in native hematopoiesis across the murine and human lifespan.bioRxiv [Preprint]. 2024 Apr 1:2024.04.01.587514. doi: 10.1101/2024.04.01.587514. bioRxiv. 2024. Update in: Nature. 2025 Jul;643(8071):478-487. doi: 10.1038/s41586-025-09041-8. PMID: 38617287 Free PMC article. Updated. Preprint.
Similar articles
-
Somatic epimutations enable single-cell lineage tracing in native hematopoiesis across the murine and human lifespan.bioRxiv [Preprint]. 2024 Apr 1:2024.04.01.587514. doi: 10.1101/2024.04.01.587514. bioRxiv. 2024. Update in: Nature. 2025 Jul;643(8071):478-487. doi: 10.1038/s41586-025-09041-8. PMID: 38617287 Free PMC article. Updated. Preprint.
-
Human brain aging is associated with dysregulation of cell type epigenetic identity.Geroscience. 2025 Jun;47(3):3759-3770. doi: 10.1007/s11357-024-01450-3. Epub 2024 Dec 27. Geroscience. 2025. PMID: 39730969 Free PMC article.
-
Mechanisms of hematopoietic clonal dominance in VEXAS syndrome.Nat Med. 2025 Jun;31(6):1911-1924. doi: 10.1038/s41591-025-03623-9. Epub 2025 Apr 7. Nat Med. 2025. PMID: 40195449 Free PMC article.
-
Exploring Epigenetic Complexity in Regulation of Hematopoietic Stem Cells Niche: A Mechanistic Journey from Normal to Malignant Hematopoiesis.Adv Exp Med Biol. 2025;1483:49-67. doi: 10.1007/5584_2024_846. Adv Exp Med Biol. 2025. PMID: 39841383 Review.
-
Stage-specific DNA methylation dynamics in mammalian heart development.Epigenomics. 2025 Apr;17(5):359-371. doi: 10.1080/17501911.2025.2467024. Epub 2025 Feb 21. Epigenomics. 2025. PMID: 39980349 Review.
Cited by
-
Personalized neoantigen vaccines in urologic malignancies.Nat Cancer. 2025 Jun;6(6):916-918. doi: 10.1038/s43018-025-00944-z. Nat Cancer. 2025. PMID: 40346293 No abstract available.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
