

Dose-dependent CHCHD10 dysregulation dictates motor neuron disease severity and alters creatine metabolism
- PMID: 40400037
- PMCID: PMC12096803
- DOI: 10.1186/s40478-025-02039-3
Dose-dependent CHCHD10 dysregulation dictates motor neuron disease severity and alters creatine metabolism
Abstract
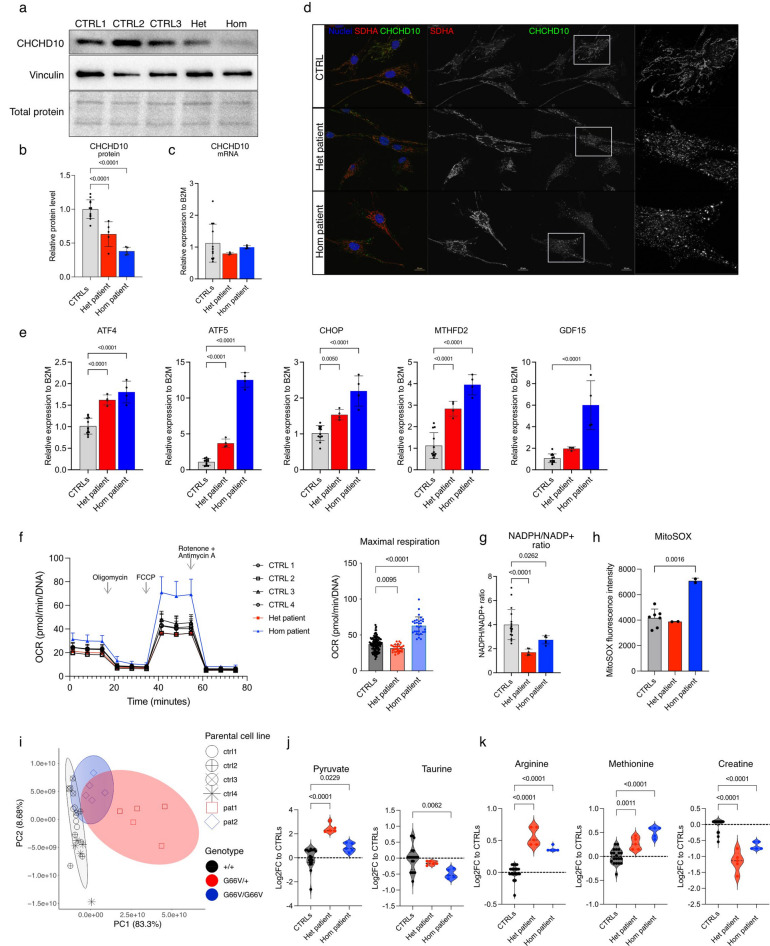
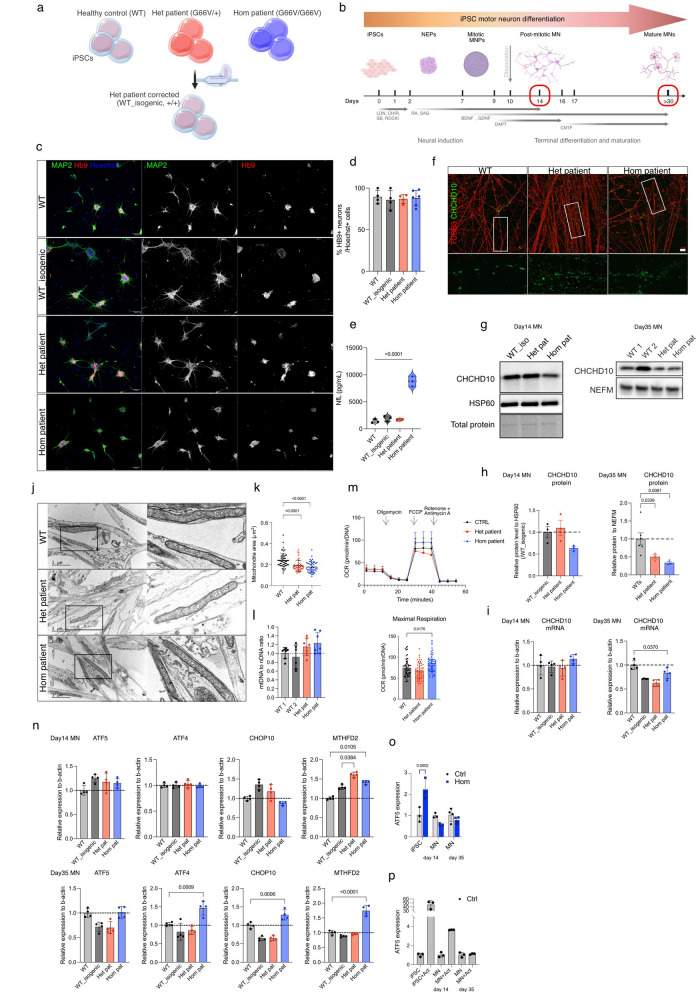
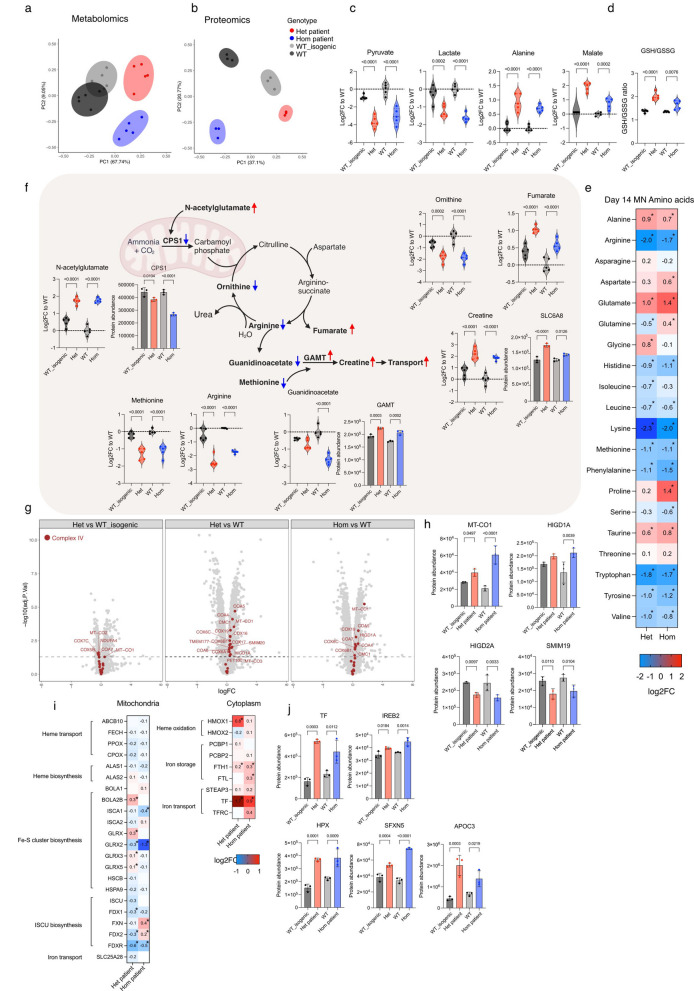
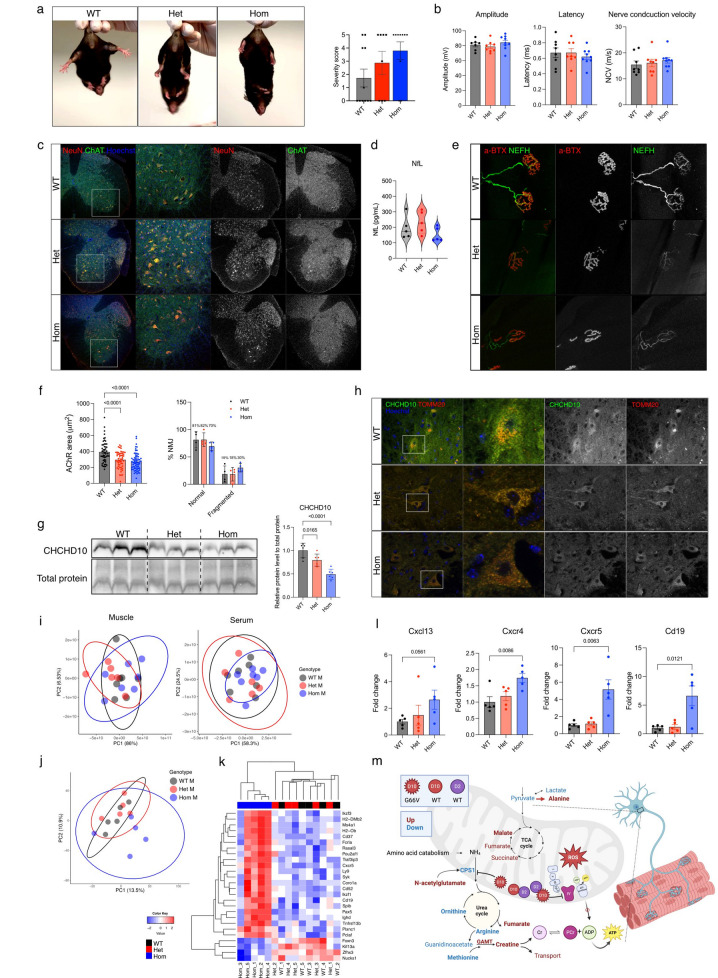
Dominant defects in CHCHD10, a mitochondrial intermembrane space protein, lead to a range of neurological and muscle disease phenotypes including amyotrophic lateral sclerosis. Many patients present with spinal muscular atrophy Jokela type (SMAJ), which is caused by heterozygous p.G66V variant. While most disease variants lead to aggregation of CHCHD10 and activation of proteotoxic stress responses, the pathogenic mechanisms of the p.G66V variant are less clear. Here we report the first homozygous CHCHD10 patient, and show that the variant dosage dictates the severity of the motor neuron disease in SMAJ. We demonstrate that the amount of the mutant CHCHD10 is reduced, but the disease mechanism of p.G66V is not full haploinsufficiency as residual mutant CHCHD10 protein is present even in a homozygous state. Novel knock-in mouse model recapitulates the dose-dependent reduction of mutant CHCHD10 protein and the slow disease progression of SMAJ. With metabolome analysis of patients' primary fibroblasts and patient-specific motor neurons, we show that CHCHD10 p.G66V dysregulates energy metabolism, leading to altered redox balance and energy buffering by creatine metabolism.
Keywords: ALS; CHCHD10; CHCHD2; Creatine; Metabolomics; Mitochondria.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Animal experiments were performed in compliance with the national ethical guidelines set by the European Union and were approved by the National Animal Experiment Board (Project Authorisation Board, project license ESAVI-12691-2021). The ethical practice of handling laboratory animals was strictly followed throughout the procedures. The generation of the human induced pluripotent stem cell lines used in this study was approved by the Coordinating Ethics Committee of the Helsinki and Uusimaa Hospital District (Nro 423/13/03/00/08) with informed consent of the donor. Consent for publication: Written and informed consent of patient was received prior examination for results and the usage of photographs for publication. Competing interests: The authors declare that they have known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. L.E. is a co-founder of NADMED Ltd. L.V.D.B. is the scientific founder of Augustine Therapeutics and is head of the Scientific Advisory Board. L.V.D.B. is also part of the Investment Advisory Board of Droia Ventures (Meise, Belgium). Authors declare no other conflicts of interest.
Figures


References
-
- Bannwarth S, Ait-El-Mkadem S, Chaussenot A, Genin EC, Lacas-Gervais S, Fragaki K, Berg-Alonso L, Kageyama Y, Serre V, Moore DG, Verschueren A, Rouzier C, Le Ber I, Augé G, Cochaud C, Lespinasse F, N’guyen K, De Septenville A, Brice A, Yu-Wai-Man P, Sesaki H, Pouget J, Paquis-Flucklinger V (2014) A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137:2329–2345. 10.1093/brain/awu138 - PMC - PubMed
-
- Chaussenot A, Le Ber I, Ait-El-Mkadem S, Camuzat A, de Septenville A, Bannwarth S, Genin EC, Serre V, Augé G, Brice A, Pouget J, Paquis-Flucklinger V (2014) Screening of CHCHD10 in a French cohort confirms the involvement of this gene in frontotemporal dementia with amyotrophic lateral sclerosis patients. Neurobiol Aging 35:2884e1–2884e4. 10.1016/j.neurobiolaging.2014.07.022 - PubMed
-
- Müller K, Andersen PM, Hübers A, Marroquin N, Volk AE, Danzer KM, Meitinger T, Ludolph AC, Strom TM, Weishaupt JH (2014) Two novel mutations in conserved codons indicate that CHCHD10 is a gene associated with motor neuron disease. Brain 137:e309 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
