

Cell simulation as cell segmentation
- PMID: 40404994
- PMCID: PMC12285883
- DOI: 10.1038/s41592-025-02697-0
Cell simulation as cell segmentation
Abstract
Single-cell spatial transcriptomics promises a highly detailed view of a cell's transcriptional state and microenvironment, yet inaccurate cell segmentation can render these data murky by misattributing large numbers of transcripts to nearby cells or conjuring nonexistent cells. We adopt methods from ab initio cell simulation, in a method called Proseg (probabilistic segmentation), to rapidly infer morphologically plausible cell boundaries. Benchmarking applied to datasets generated by three commercial platforms shows superior performance and computational efficiency of Proseg when compared to existing methods. We show that improved accuracy in cell segmentation aids greatly in detection of difficult-to-segment tumor-infiltrating immune cells such as neutrophils and T cells. Last, through improvements in our ability to delineate subsets of tumor-infiltrating T cells, we show that CXCL13-expressing CD8+ T cells tend to be more closely associated with tumor cells than their CXCL13-negative counterparts in data generated from samples from patients with renal cell carcinoma.
© 2025. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests: E.W.N. is a co-founder, advisor and shareholder of ImmunoScape and is an advisor for Neogene Therapuetics and Nanostring Technologies. D.C.J. is listed as the inventor in a patent application for methods implemented in Proseg, submitted by the Fred Hutchinson Cancer Center. The other authors declare no competing interests.
Figures










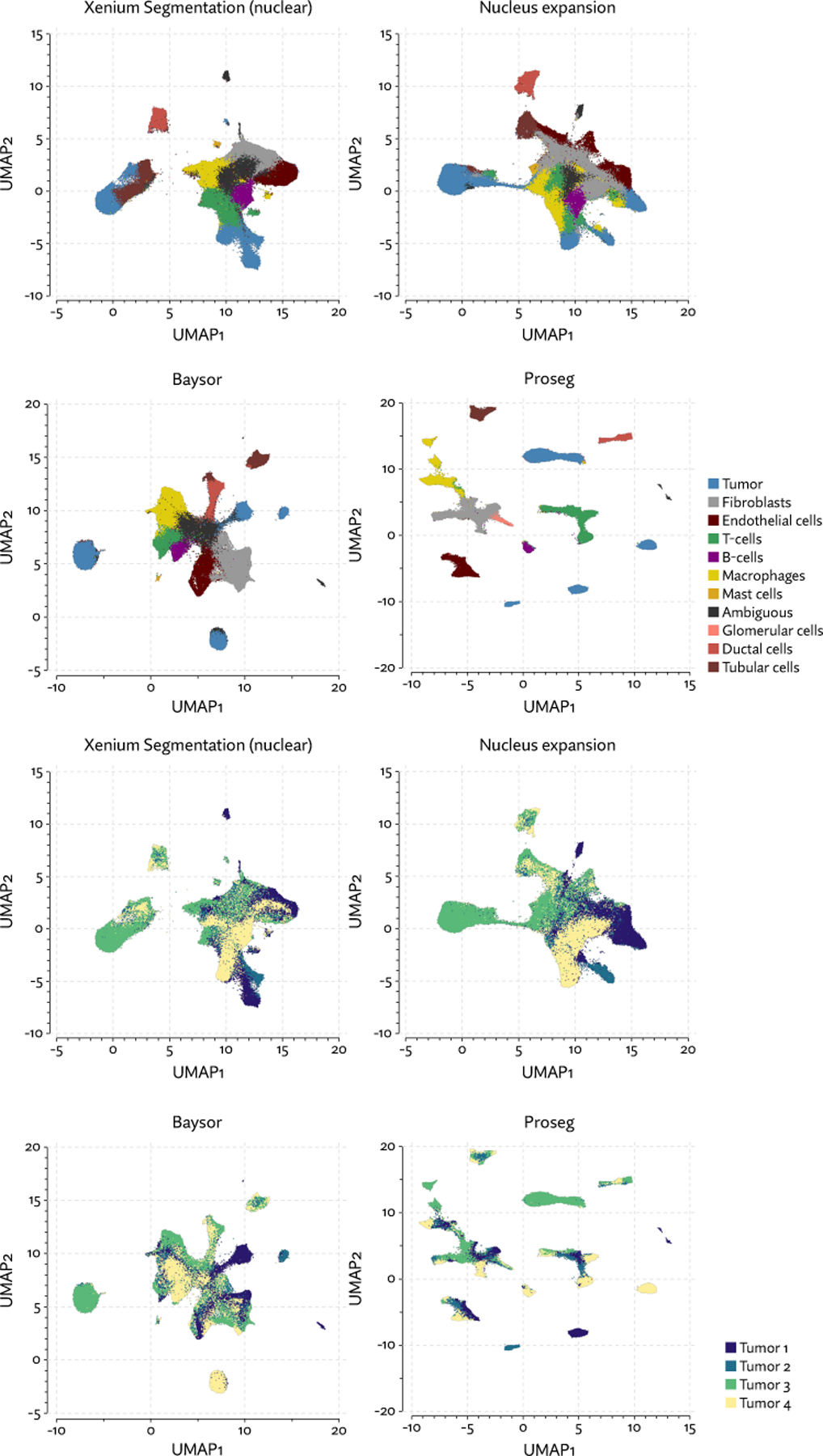

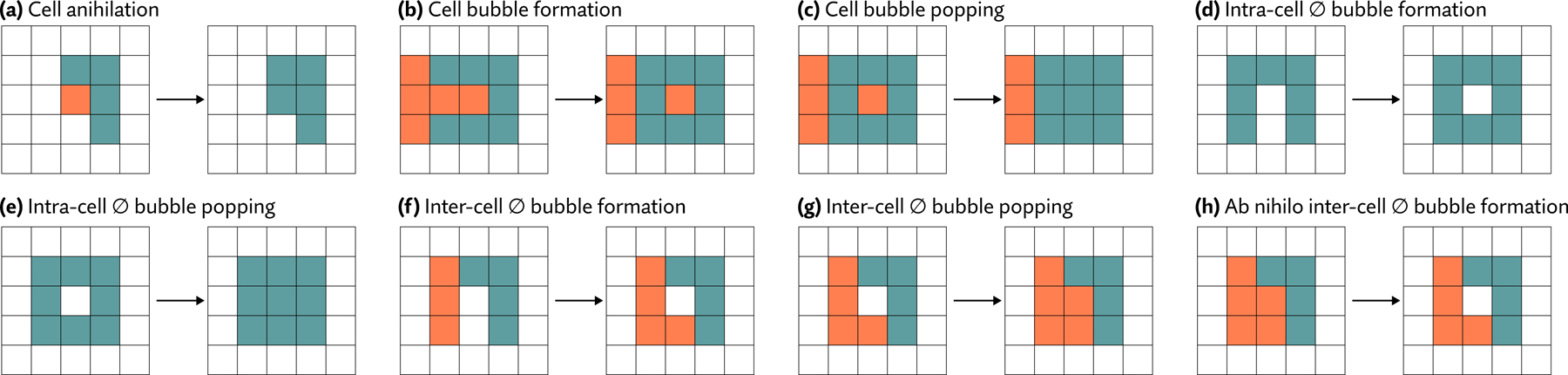
Update of
-
Cell Simulation as Cell Segmentation.bioRxiv [Preprint]. 2024 Jul 9:2024.04.25.591218. doi: 10.1101/2024.04.25.591218. bioRxiv. 2024. Update in: Nat Methods. 2025 Jun;22(6):1331-1342. doi: 10.1038/s41592-025-02697-0. PMID: 38712065 Free PMC article. Updated. Preprint.
References
-
- He Shanshan, Bhatt Ruchir, Brown Carl, Brown Emily A, Buhr Derek L, Chantranuvatana Kan, Danaher Patrick, Dunaway Dwayne, Garrison Ryan G, Geiss Gary, Gregory Mark T, Hoang Margaret L, Khafizov Rustem, Killing-beck Emily E, Kim Dae, Kim Tae Kyung, Kim Youngmi, Klock Andrew, Korukonda Mithra, Kutchma Alecksandr, Lee Erica, Lewis Zachary R, Liang Yan, Nelson Jeffrey S, Ong Giang T, Perillo Evan P, Phan Joseph C, Phan-Everson Tien, Piazza Erin, Rane Tushar, Reitz Zachary, Rhodes Michael, Rosenbloom Alyssa, Ross David, Sato Hiromi, Wardhani Aster W, Williams-Wietzikoski Corey A, Wu Lidan, and Beechem Joseph M. High-plex multiomic analysis in FFPE at subcellular level by spatial molecular imaging. bioRxiv, page 2021.11.03.467020, January 2022.
-
- Janesick Amanda, Shelansky Robert, Gottscho Andrew, Wagner Florian, Rouault Morgane, Beliakoff Ghezal, Oliveira Michelli Faria de, Kohlway Andrew, Abousoud Jawad, Morrison Carolyn, Drennon Tingsheng Yu, Mohabbat Syrus, Williams Stephen, and Taylor Sarah. High resolution mapping of the breast cancer tumor microenvironment using integrated single cell, spatial and in situ analysis of FFPE tissue. bioRxiv, page 2022.10.06.510405, October 2022.
-
- Beucher S and Meyer F. The morphological approach to segmentation: The watershed transformation. In Mathematical Morphology in Image Processing, pages 433–481. CRC Press, 1992.
-
- Ronneberger Olaf, Fischer Philipp, and Brox Thomas. U-net: Convolutional networks for biomedical image segmentation. arXiv [cs.CV], May 2015.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
