

Tumor antigens preferentially derive from unmutated genomic sequences in melanoma and non-small cell lung cancer
- PMID: 40405018
- PMCID: PMC12380612
- DOI: 10.1038/s43018-025-00979-2
Tumor antigens preferentially derive from unmutated genomic sequences in melanoma and non-small cell lung cancer
Abstract
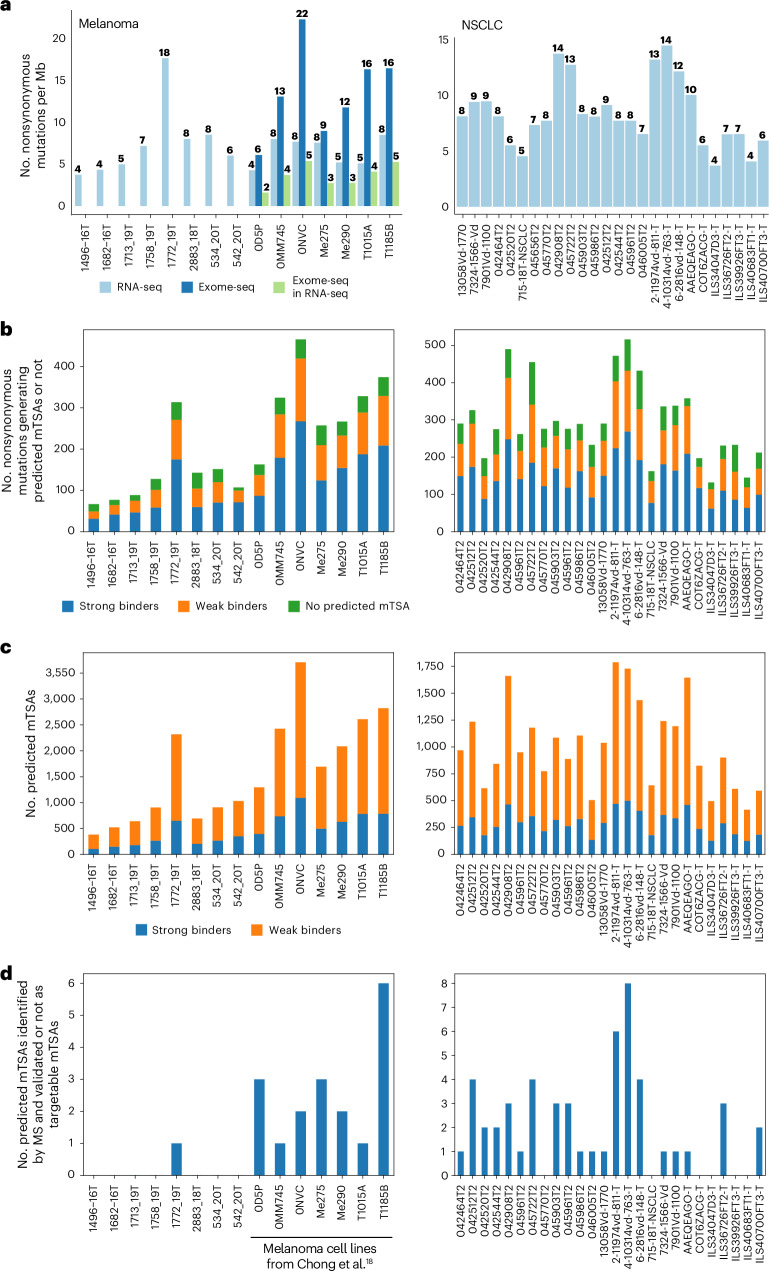
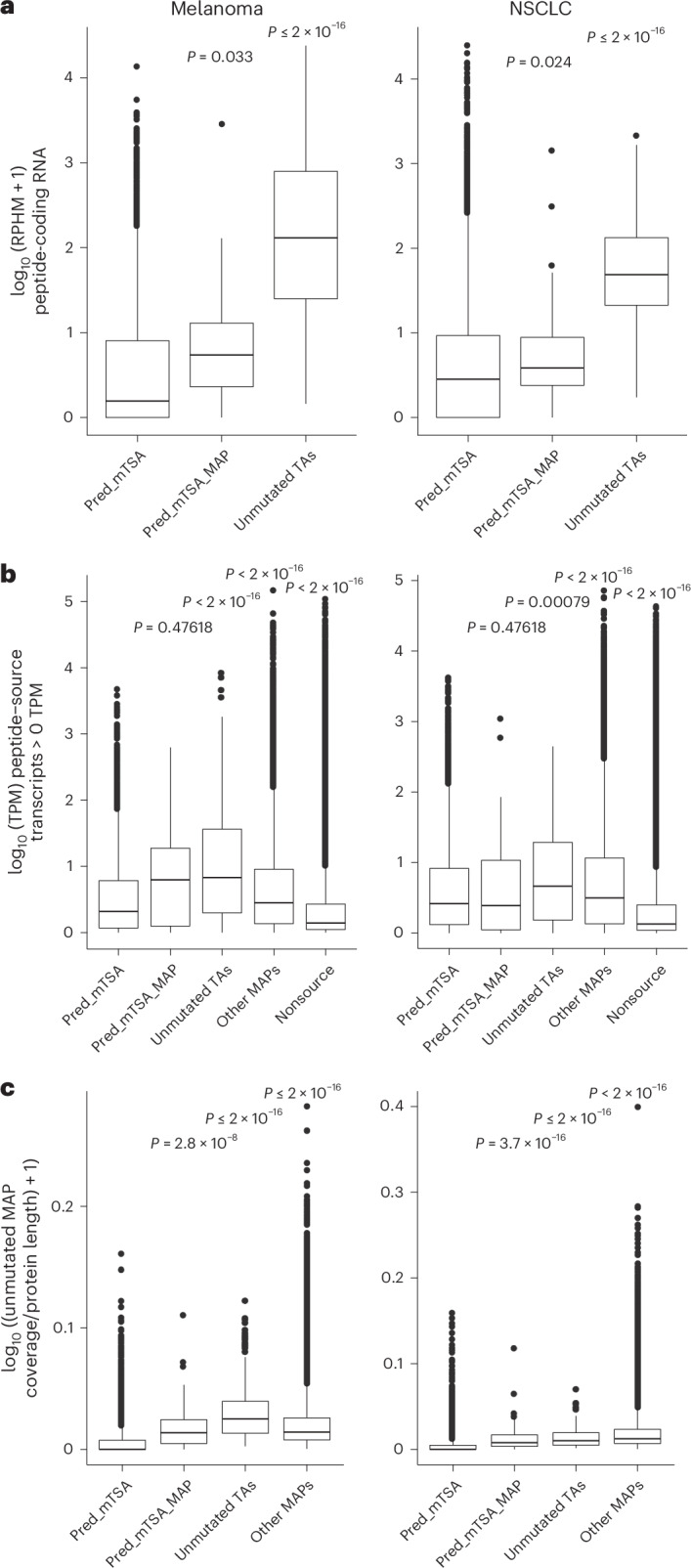
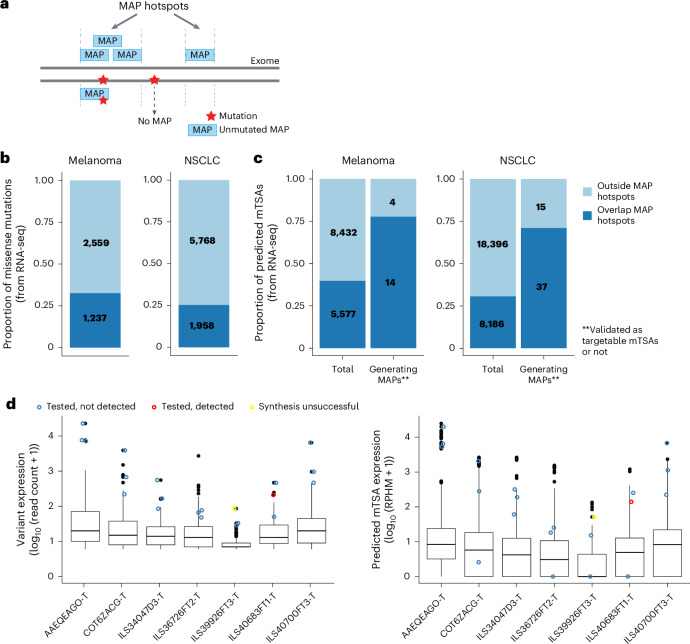
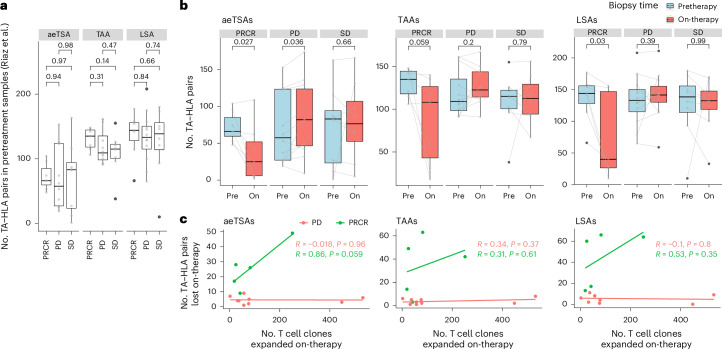
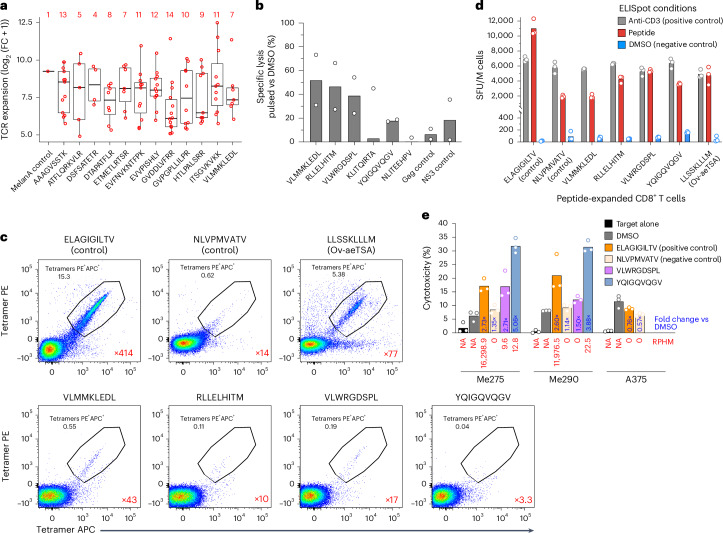
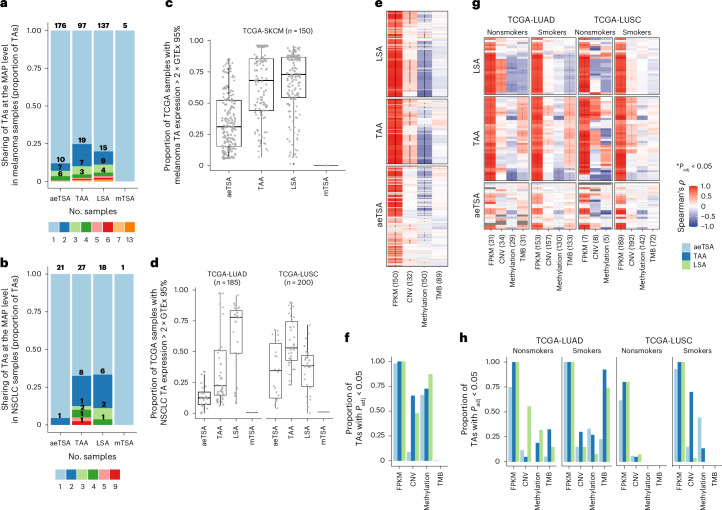
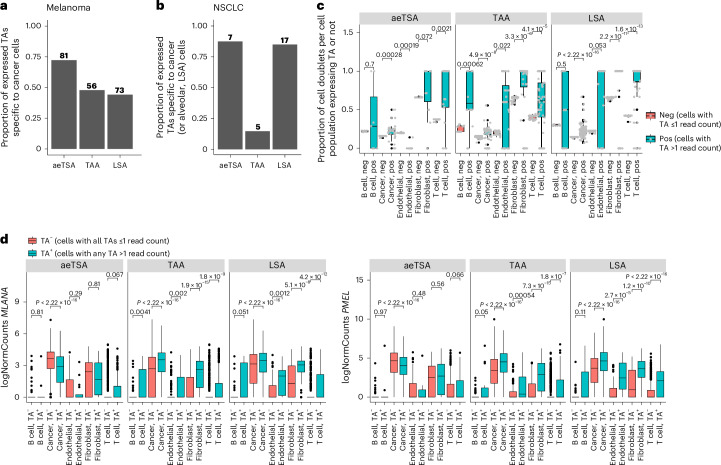
Melanoma and non-small cell lung cancer (NSCLC) display exceptionally high mutational burdens. Hence, immune targeting in these cancers has primarily focused on tumor antigens (TAs) predicted to derive from nonsynonymous mutations. Using comprehensive proteogenomic analyses, we identified 589 TAs in cutaneous melanoma (n = 505) and NSCLC (n = 90). Of these, only 1% were derived from mutated sequences, which was explained by a low RNA expression of most nonsynonymous mutations and their localization outside genomic regions proficient for major histocompatibility complex (MHC) class I-associated peptide generation. By contrast, 99% of TAs originated from unmutated genomic sequences specific to cancer (aberrantly expressed tumor-specific antigens (aeTSAs), n = 220), overexpressed in cancer (tumor-associated antigens (TAAs), n = 165) or specific to the cell lineage of origin (lineage-specific antigens (LSAs), n = 198). Expression of aeTSAs was epigenetically regulated, and most were encoded by noncanonical genomic sequences. aeTSAs were shared among tumor samples, were immunogenic and could contribute to the response to immune checkpoint blockade observed in previous studies, supporting their immune targeting across cancers.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: A.A., K.V., M.-P.H., P.T. and C.P. are named inventors on patent applications filed by the Université de Montréal and covering TAs reported in this article (patent application number WO2024211992, titled Novel tumor antigens for melanoma and uses thereof; patent application number WO2024187278, titled Novel tumor antigens for lung cancer and uses thereof). P.T. and C.P. receive grant support and consultant fees from Epitopea. The other authors declare no competing interests.
Figures











References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
