

Intrinsic Disorder and Phase Separation Coordinate Exocytosis, Motility, and Chromatin Remodeling in the Human Acrosomal Proteome
- PMID: 40407495
- PMCID: PMC12101322
- DOI: 10.3390/proteomes13020016
Intrinsic Disorder and Phase Separation Coordinate Exocytosis, Motility, and Chromatin Remodeling in the Human Acrosomal Proteome
Abstract
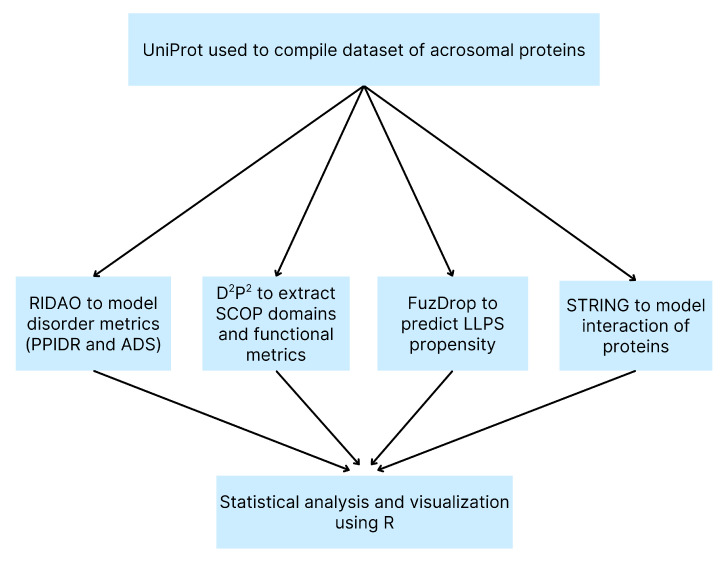
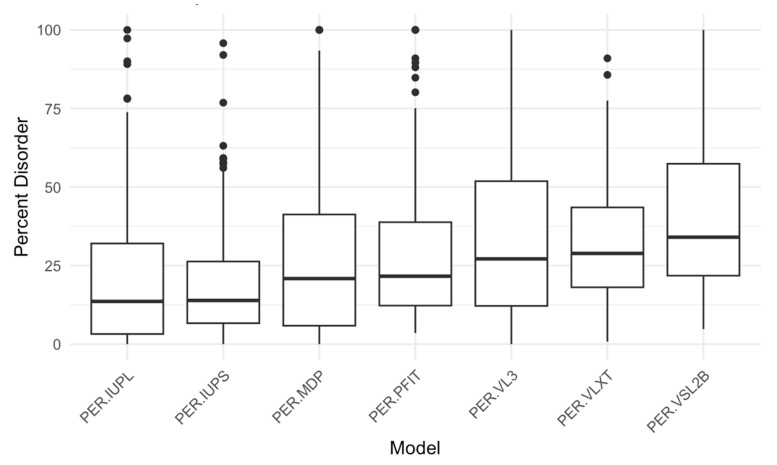
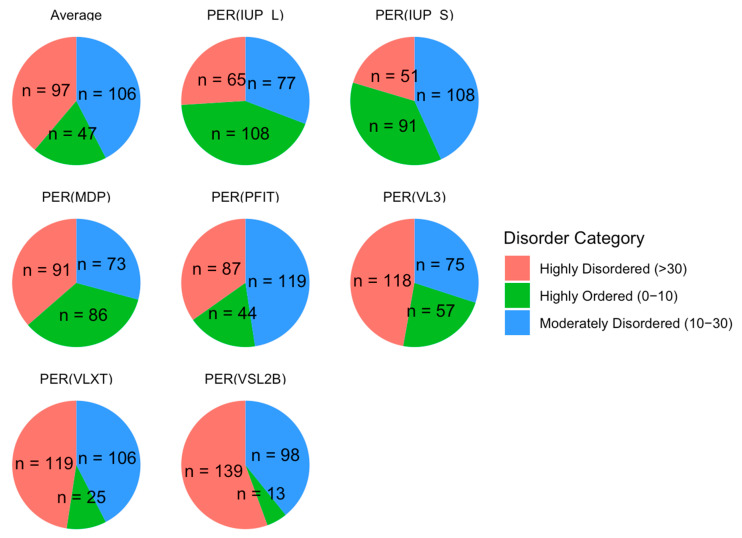
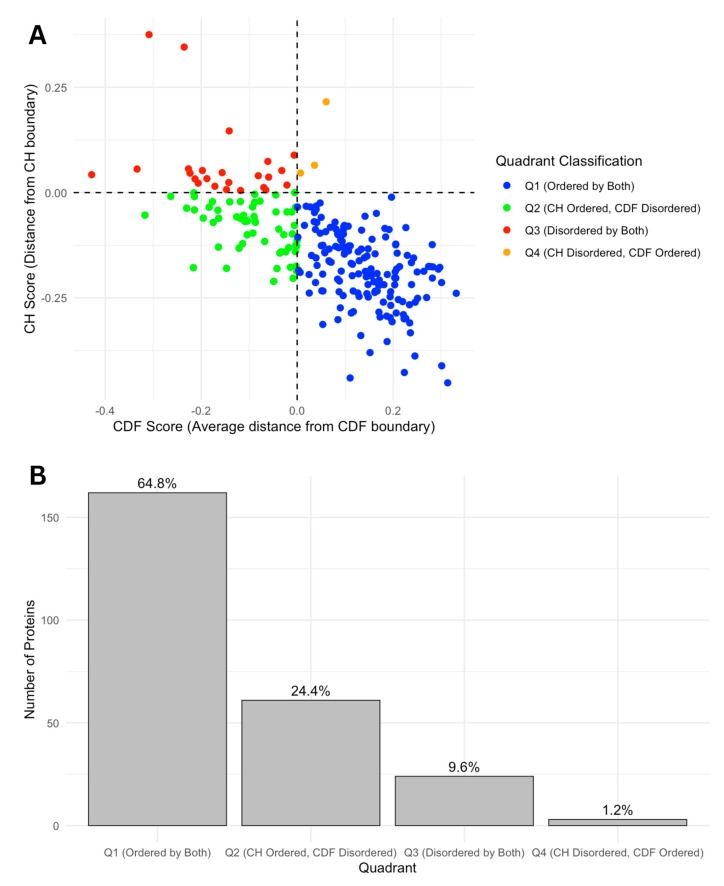
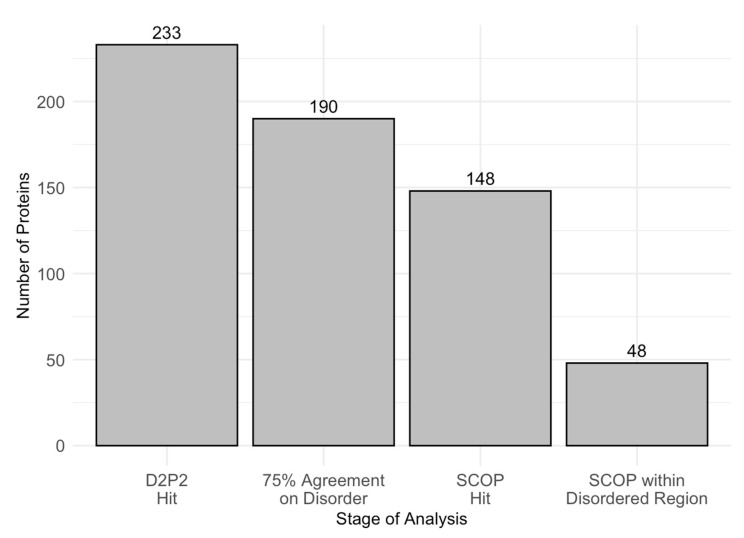
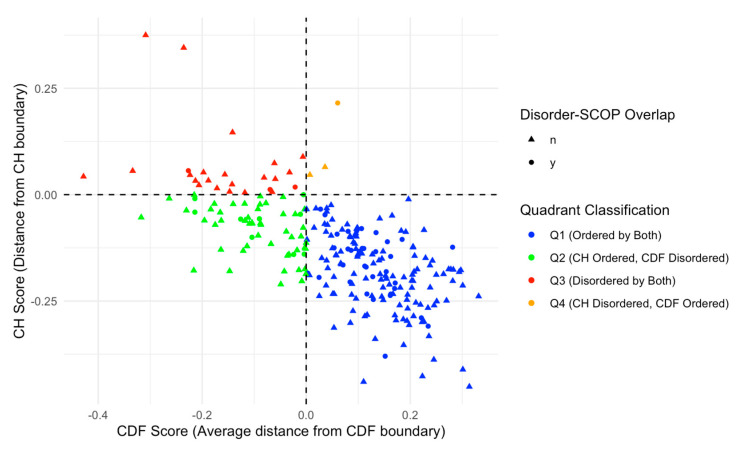
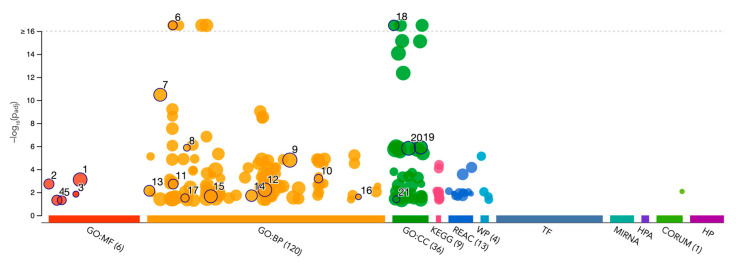
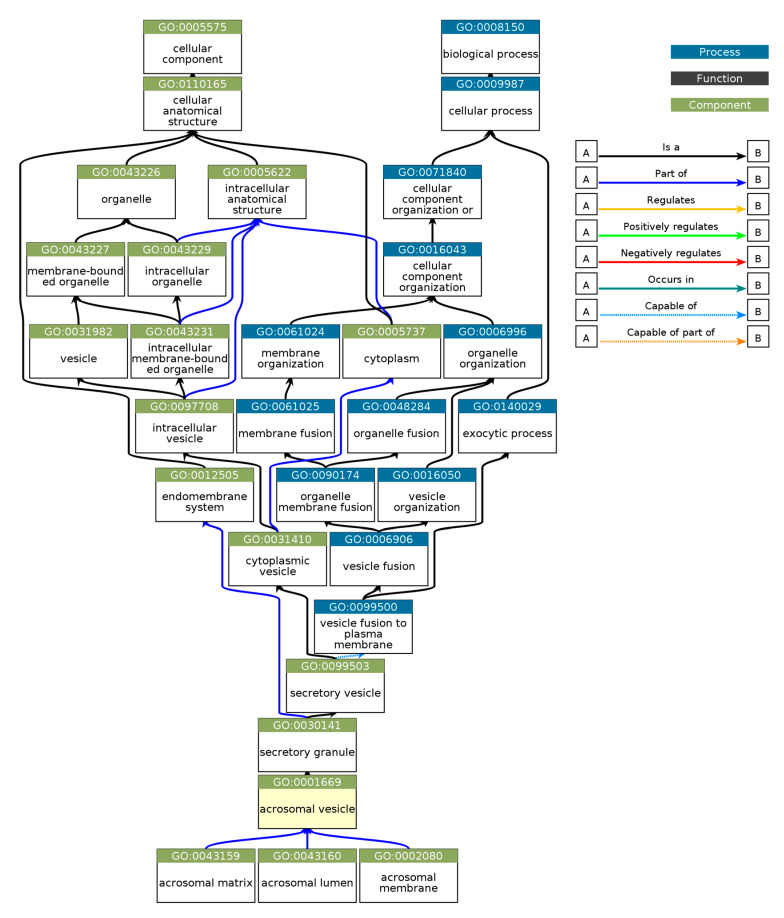
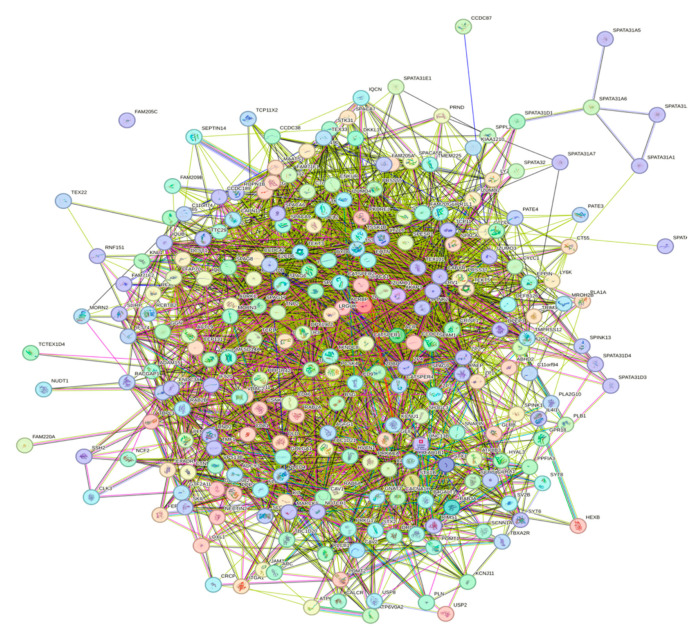
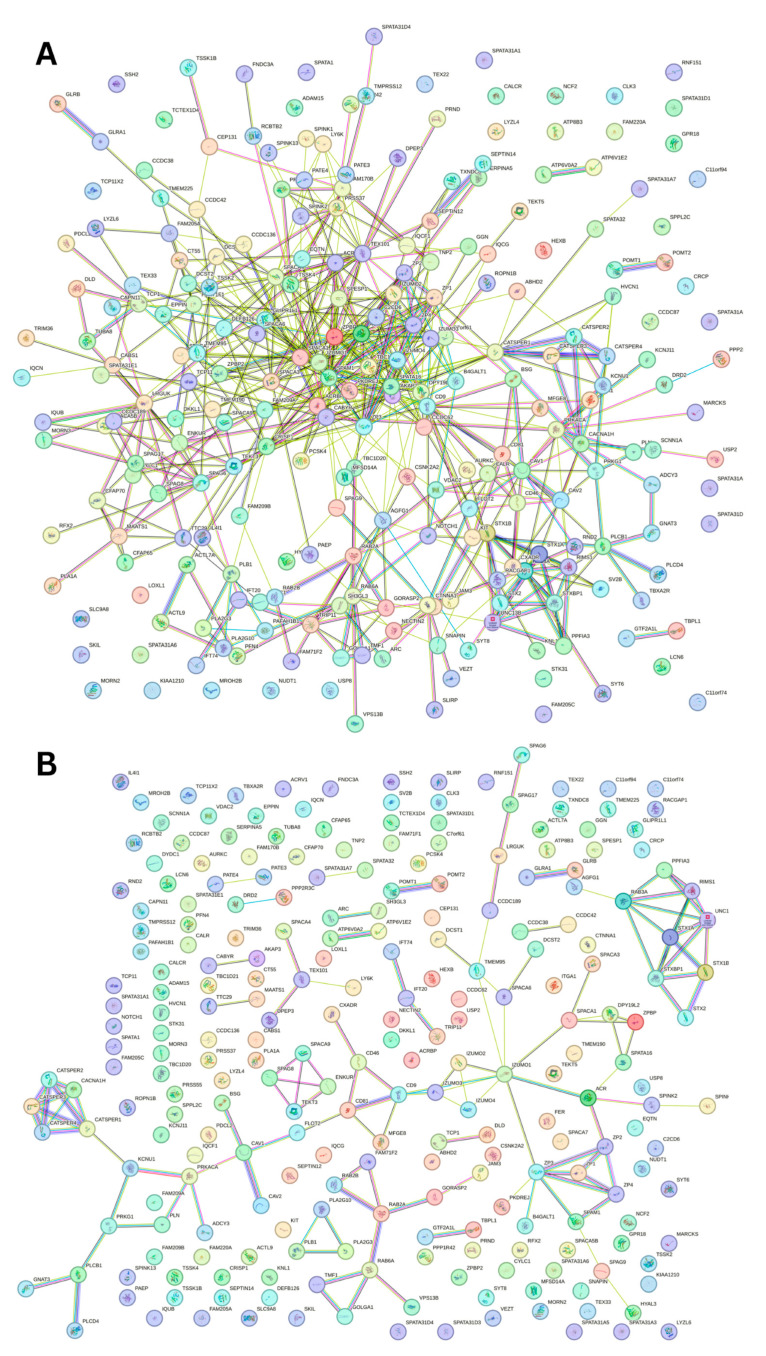
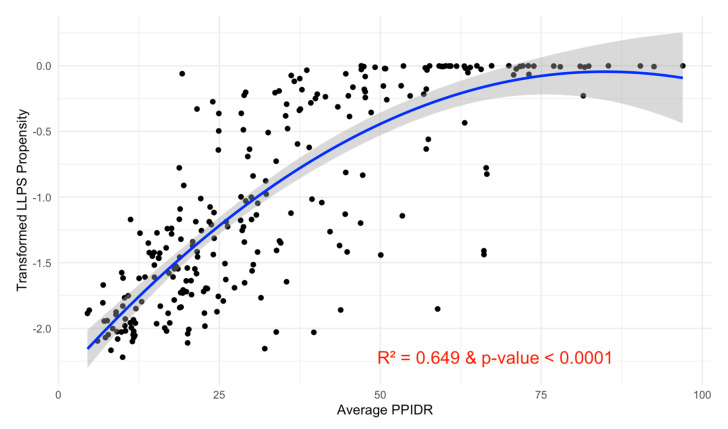
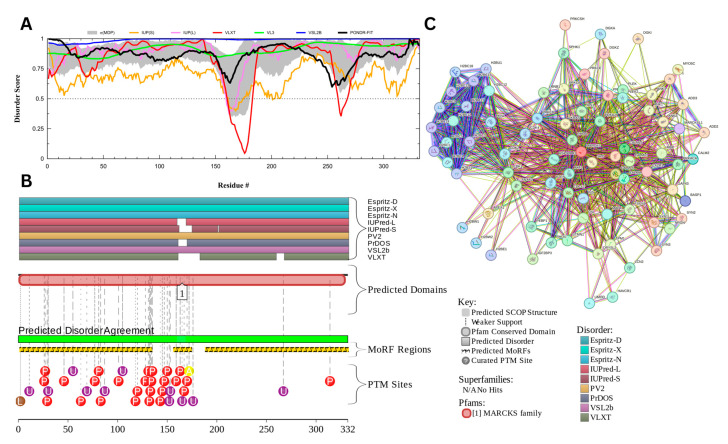
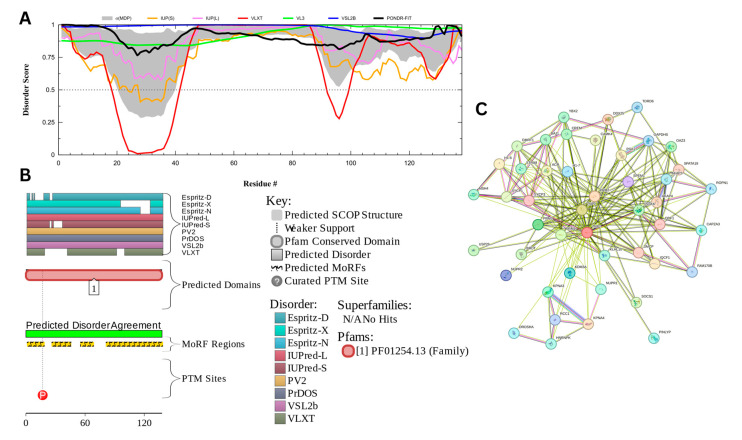
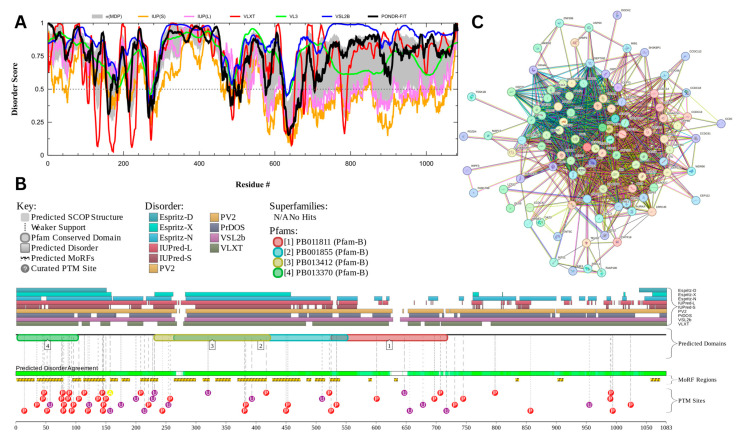
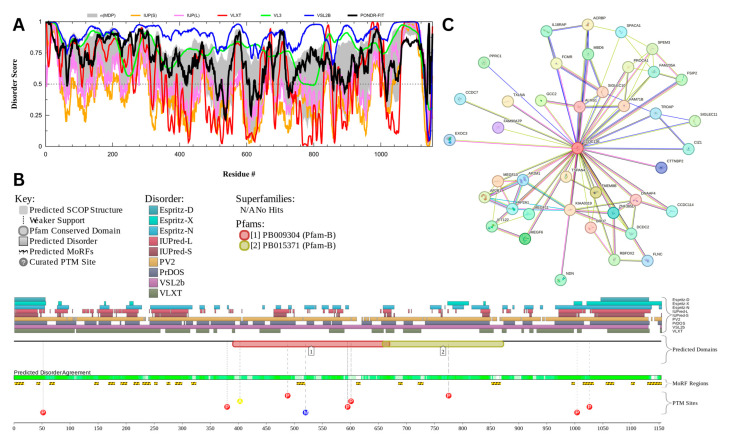
Intrinsic disorder refers to protein regions that lack a fixed three-dimensional structure under physiological conditions, enabling conformational plasticity. This flexibility allows for diverse functions, including transient interactions, signaling, and phase separation via disorder-to-order transitions upon binding. Our study focused on investigating the role of intrinsic disorder and liquid-liquid phase separation (LLPS) in the human acrosome, a sperm-specific organelle essential for fertilization. Using computational prediction models, network analysis, Structural Classification of Proteins (SCOP) functional assessments, and Gene Ontology, we analyzed 250 proteins within the acrosomal proteome. Our bioinformatic analysis yielded 97 proteins with high levels (>30%) of structural disorder. Further analysis of functional enrichment identified associations between disordered regions overlapping with SCOP domains and critical acrosomal processes, including vesicle trafficking, membrane fusion, and enzymatic activation. Examples of disordered SCOP domains include the PLC-like phosphodiesterase domain, the t-SNARE domain, and the P-domain of calnexin/calreticulin. Protein-protein interaction networks revealed acrosomal proteins as hubs in tightly interconnected systems, emphasizing their functional importance. LLPS propensity modeling determined that over 30% of these proteins are high-probability LLPS drivers (>60%), underscoring their role in dynamic compartmentalization. Proteins such as myristoylated alanine-rich C-kinase substrate and nuclear transition protein 2 exhibited both high LLPS propensities and high levels of structural disorder. A significant relationship (p < 0.0001, R² = 0.649) was observed between the level of intrinsic disorder and LLPS propensity, showing the role of disorder in facilitating phase separation. Overall, these findings provide insights into how intrinsic disorder and LLPS contribute to the structural adaptability and functional precision required for fertilization, with implications for understanding disorders associated with the human acrosome reaction.
Keywords: acrosomal proteins; acrosome; bioinformatics; fertility; human sperm; intrinsic disorder; proteins; reproduction; spermatogenesis; structure.
Conflict of interest statement
The authors declare no conflicts of interest. The funders had no role in the design of this study; in the collection, analyses, or interpretation of data; in the writing of this manuscript; or in the decision to publish the results.
Figures



Similar articles
-
An Analysis of Intrinsic Protein Disorder in Antimicrobial Peptides.Protein J. 2025 Apr;44(2):175-191. doi: 10.1007/s10930-025-10253-0. Epub 2025 Feb 20. Protein J. 2025. PMID: 39979561 Free PMC article.
-
Comprehensive analysis of liquid-liquid phase separation propensities of HSV-1 proteins and their interaction with host factors.J Cell Biochem. 2024 Dec;125(12):e30480. doi: 10.1002/jcb.30480. Epub 2023 Oct 5. J Cell Biochem. 2024. PMID: 37796176
-
Characterization of a novel role for the dynamin mechanoenzymes in the regulation of human sperm acrosomal exocytosis.Mol Hum Reprod. 2017 Oct 1;23(10):657-673. doi: 10.1093/molehr/gax044. Mol Hum Reprod. 2017. PMID: 29044420
-
The Role of Zinc in Male Fertility.Int J Mol Sci. 2020 Oct 21;21(20):7796. doi: 10.3390/ijms21207796. Int J Mol Sci. 2020. PMID: 33096823 Free PMC article. Review.
-
The (un)structural biology of biomolecular liquid-liquid phase separation using NMR spectroscopy.J Biol Chem. 2020 Feb 21;295(8):2375-2384. doi: 10.1074/jbc.REV119.009847. Epub 2020 Jan 7. J Biol Chem. 2020. PMID: 31911439 Free PMC article. Review.
References
-
- Erenpreisa J., Vainshelbaum N.M., Lazovska M., Karklins R., Salmina K., Zayakin P., Rumnieks F., Inashkina I., Pjanova D., Erenpreiss J. The Price of Human Evolution: Cancer-Testis Antigens, the Decline in Male Fertility and the Increase in Cancer. Int. J. Mol. Sci. 2023;24:11660. doi: 10.3390/ijms241411660. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
