

Interplay of kinetochores and catalysts drives rapid assembly of the mitotic checkpoint complex
- PMID: 40410156
- PMCID: PMC12102207
- DOI: 10.1038/s41467-025-59970-1
Interplay of kinetochores and catalysts drives rapid assembly of the mitotic checkpoint complex
Abstract
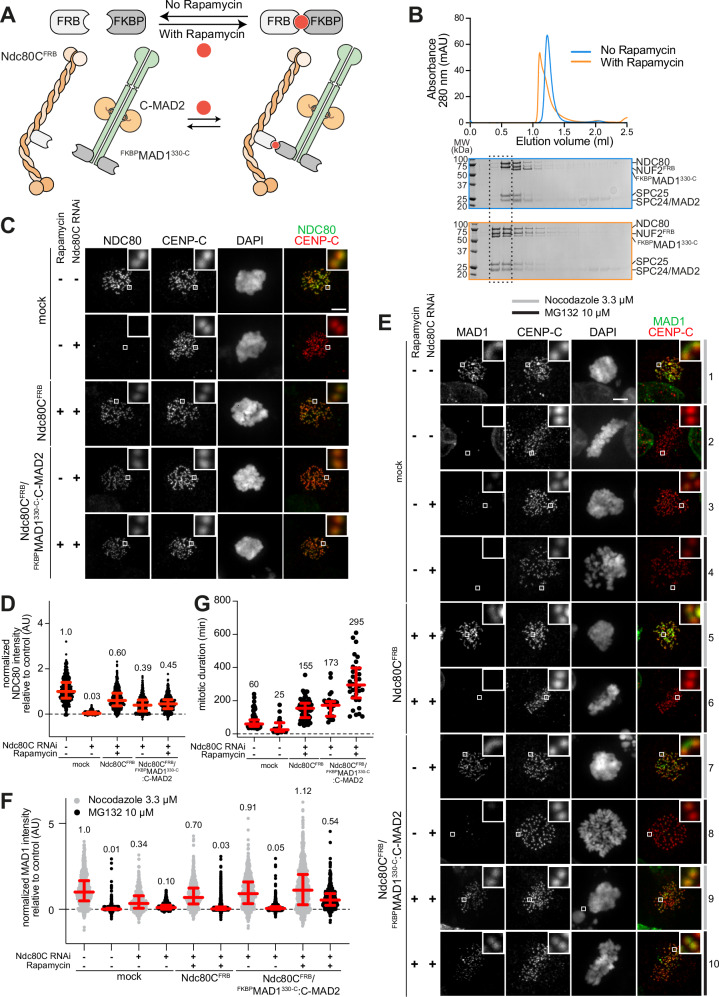
The spindle assembly checkpoint (SAC) ensures mitotic exit occurs only after sister chromatid biorientation, but how this coordination is mechanistically achieved remains unclear. Kinetochores, the megadalton complexes linking chromosomes to spindle microtubules, contribute to SAC signaling. However, whether they act solely as docking platforms or actively promote the co-orientation of SAC catalysts such as MAD1:MAD2 and BUB1:BUB3 remains unresolved. Here, we reconstitute kinetochores and SAC signaling in vitro to address this question. We engineer recombinant kinetochore particles that recruit core SAC components and trigger checkpoint signaling upon Rapamycin induction, and test their function using a panel of targeted mutants. At approximately physiological concentrations of SAC proteins, kinetochores are essential for efficient mitotic checkpoint complex (MCC) assembly, the key effector of SAC signaling. Our results suggest that kinetochores serve not only as structural hubs but also as catalytic platforms that concentrate and spatially organize SAC components to accelerate MCC formation and ensure timely checkpoint activation.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures






Similar articles
-
KNL1-Bubs and RZZ Provide Two Separable Pathways for Checkpoint Activation at Human Kinetochores.Dev Cell. 2015 Dec 7;35(5):600-613. doi: 10.1016/j.devcel.2015.11.012. Dev Cell. 2015. PMID: 26651294
-
Basis of catalytic assembly of the mitotic checkpoint complex.Nature. 2017 Feb 23;542(7642):498-502. doi: 10.1038/nature21384. Epub 2017 Jan 19. Nature. 2017. PMID: 28102834 Free PMC article.
-
The structural flexibility of MAD1 facilitates the assembly of the Mitotic Checkpoint Complex.Nat Commun. 2023 Mar 18;14(1):1529. doi: 10.1038/s41467-023-37235-z. Nat Commun. 2023. PMID: 36934097 Free PMC article.
-
Orchestration of the spindle assembly checkpoint by CDK1-cyclin B1.FEBS Lett. 2019 Oct;593(20):2889-2907. doi: 10.1002/1873-3468.13591. Epub 2019 Sep 13. FEBS Lett. 2019. PMID: 31469407 Review.
-
How the SAC gets the axe: Integrating kinetochore microtubule attachments with spindle assembly checkpoint signaling.Bioarchitecture. 2015;5(1-2):1-12. doi: 10.1080/19490992.2015.1090669. Epub 2015 Oct 2. Bioarchitecture. 2015. PMID: 26430805 Free PMC article. Review.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
