

Angiopoietin-TIE2 feedforward circuit promotes PIK3CA-driven venous malformations
- PMID: 40410415
- PMCID: PMC12259471
- DOI: 10.1038/s44161-025-00655-9
Angiopoietin-TIE2 feedforward circuit promotes PIK3CA-driven venous malformations
Abstract
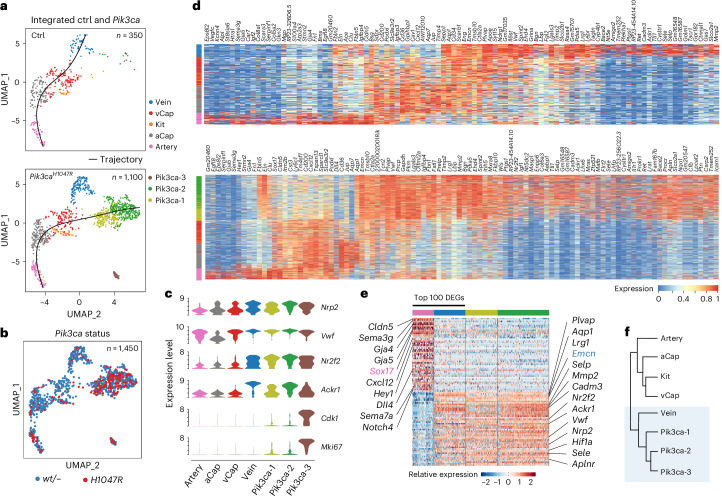
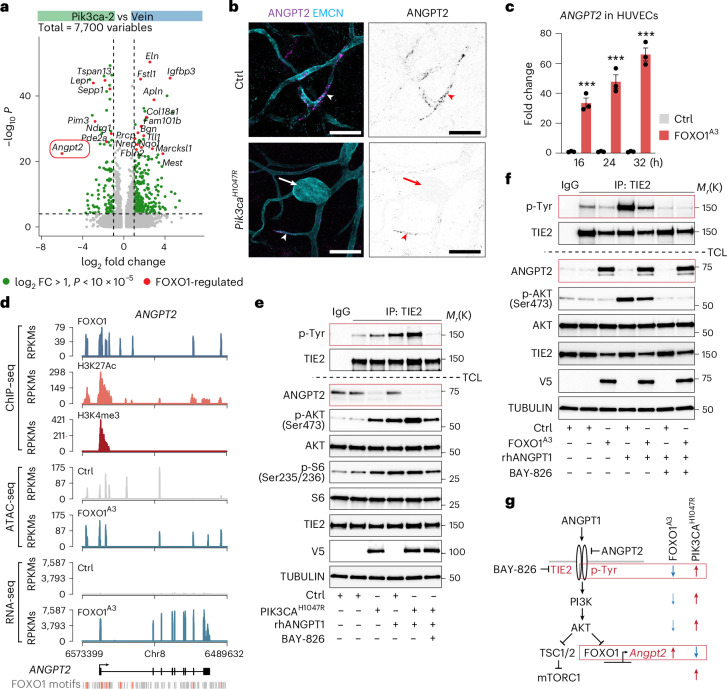
Venous malformations (VMs) are vascular anomalies lacking curative treatments, often caused by somatic PIK3CA mutations that hyperactivate the PI3Kα-AKT-mTOR signaling pathway. Here, we identify a venous-specific signaling circuit driving disease progression, where excessive PI3Kα activity amplifies upstream TIE2 receptor signaling through autocrine and paracrine mechanisms. In Pik3caH1047R-driven VM mouse models, single-cell transcriptomics and lineage tracking revealed clonal expansion of mutant endothelial cells with a post-capillary venous phenotype, characterized by suppression of the AKT-inhibited FOXO1 and its target genes, including the TIE2 antagonist ANGPT2. An imbalance in TIE2 ligands, likely exacerbated by aberrant recruitment of smooth muscle cells producing the agonist ANGPT1, increased TIE2 activity in both mouse and human VMs. While mTOR blockade had limited effects on advanced VMs in mice, inhibiting TIE2 or ANGPT effectively suppressed their growth. These findings uncover a PI3K-FOXO1-ANGPT-TIE2 circuit as a core driver of PIK3CA-related VMs and highlight TIE2 as a promising therapeutic target.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
















References
MeSH terms
Substances
Grants and funding
- 2018.0218/Knut och Alice Wallenbergs Stiftelse (Knut and Alice Wallenberg Foundation)
- 2020.0057/Knut och Alice Wallenbergs Stiftelse (Knut and Alice Wallenberg Foundation)
- 2020-0269/Vetenskapsrådet (Swedish Research Council)
- 240137/Sigrid Juséliuksen Säätiö (Sigrid Jusélius Foundation)
- 814316/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 Marie Sklodowska-Curie Actions (H2020 Excellent Science - Marie Sklodowska-Curie Actions)
- 19 0220 Pj, 22 2025 Pj/Cancerfonden (Swedish Cancer Society)
- T.00.19.22 and P.C013.20/Fonds De La Recherche Scientifique - FNRS (Belgian National Fund for Scientific Research)
- T.0240.23 & P.C005.22/Fonds De La Recherche Scientifique - FNRS (Belgian National Fund for Scientific Research)
- 101078827/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European Research Council)
- 773047/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European Research Council)
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous