

Multi-Omics and -Organ Insights into Energy Metabolic Adaptations in Early Sepsis Onset
- PMID: 40411399
- PMCID: PMC12376650
- DOI: 10.1002/advs.202504418
Multi-Omics and -Organ Insights into Energy Metabolic Adaptations in Early Sepsis Onset
Abstract
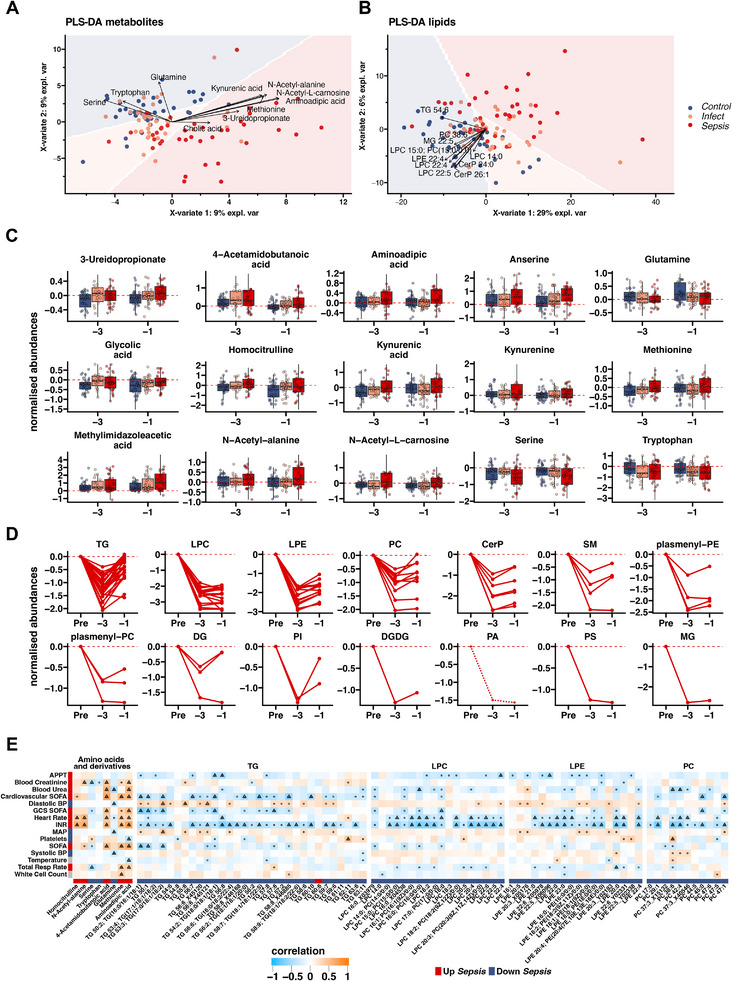
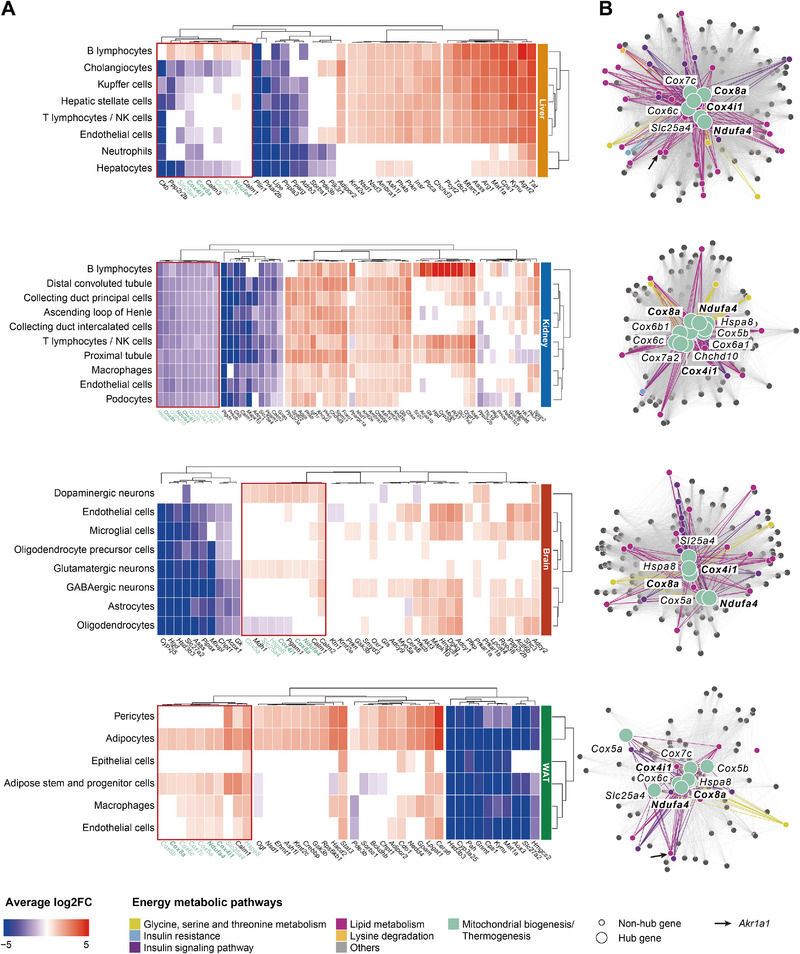
Systemic metabolic dysregulation in sepsis critically impacts patient survival. To better understand its onset, untargeted serum metabolomics and lipidomics are analyzed from 152 presymptomatic patients undergoing major elective surgery, and identified key metabolites, including serine and aminoadipic acid, that differentiate postoperative uncomplicated infection from sepsis. Using single-nucleus RNA sequencing data from an in vivo mouse model of sepsis, tissue-independent down-regulation and tissue-specific differences of serine and energy-related genes including key module roles for the mitochondria-linked genes, Cox4i1, Cox8a, and Ndufa4 are identified. Finally, serine-dependent metabolic shifts, especially in the liver, are revealed by using 12C/13C murine data with labeled serine, and link altered activity of the serine hydroxymethyltransferase (SHMT) cycle with perturbed purine metabolism during sepsis. This study demonstrates the close interrelationship between early metabolite changes and mitochondrial dysfunction in sepsis, improves the understanding of the underlying pathophysiology, and highlights metabolic targets to prospectively treat presymptomatic, but at-risk, patients.
Keywords: cell–cell communication; energy imbalance; lipid metabolism; metabolic modeling; sepsis biomarkers; single‐nucleus RNA sequencing (snRNA‐Seq).
© 2025 The Author(s). Advanced Science published by Wiley‐VCH GmbH.
Conflict of interest statement
M.S. received grants from NewB, Apollo Therapeutics, and UCL Technology Fund and others from Abbott, Amormed, bioMérieux, Biotest, Deltex Medical, Fresenius, Mindray, NewB, Pfizer, Radiometer, Roche Diagnostics, Safeguard Biosystems, Shionogi, and Spiden outside of this project. M.S. is an unpaid advisor to Presymptom Health Ltd, hemotune, deepUll, and Santersus, and R.L. is a part‐time advisor to Presymptom Health Ltd.
Figures





References
-
- Singer M., Deutschman C. S., Seymour C., Shankar‐Hari M., Annane D., Bauer M., Bellomo R., Bernard G. R., Chiche J. D., Coopersmith C. M., Hotchkiss R. S., Levy M. M., Marshall J. C., Martin G. S., Opal S. M., Rubenfeld G. D., Poll T. D., Vincent J. L., Angus D. C., J. Am. Med. Assoc. 2016, 315, 801.
-
- Rudd K. E., Johnson S. C., Agesa K. M., Shackelford K. A., Tsoi D., Kievlan D. R., Colombara D. V., Ikuta K. S., Kissoon N., Finfer S., Fleischmann‐Struzek C., Machado F. R., Reinhart K. K., Rowan K., Seymour C. W., Watson R. S., West T. E., Marinho F., Hay S. I., Lozano R., Lopez A. D., Angus D. C., Murray C. J. L., Naghavi M., Lancet 2020, 395, 200. - PMC - PubMed
-
- Evans L., Rhodes A., Alhazzani W., Antonelli M., Coopersmith C. M., French C., Machado F. R., Mcintyre L., Ostermann M., Prescott H. C., Schorr C., Simpson S., Wiersinga W. J., Alshamsi F., Angus D. C., Arabi Y., Azevedo L., Beale R., Beilman G., Belley‐Cote E., Burry L., Cecconi M., Centofanti J., Coz Yataco A., De Waele J., Dellinger R. P., Doi K., Du B., Estenssoro E., Ferrer R., et al., Intens. Care Med. 2021, 47, 1181. - PMC - PubMed
-
- Giamarellos‐Bourboulis E. J., Aschenbrenner A. C., Bauer M., Bock C., Calandra T., Gat‐Viks I., Kyriazopoulou E., Lupse M., Monneret G., Pickkers P., Schultze J. L., van der Poll T., van de Veerdonk F. L., Vlaar A. P. J., Weis S., Wiersinga W. J., Netea M. G., Nat. Immunol. 2024, 25, 19. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical