

Evaluation of aggrephagy markers in myofibrillar myopathies
- PMID: 40413523
- PMCID: PMC12102939
- DOI: 10.1186/s40478-025-02041-9
Evaluation of aggrephagy markers in myofibrillar myopathies
Abstract
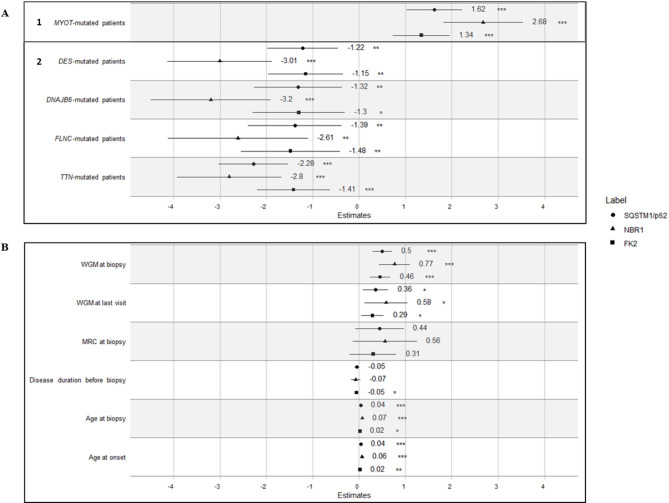
Myofibrillar Myopathies (MFMs) are a growing group of muscular disorders genetically determined, whose diagnosis is based on histological features as myofibrillar degeneration, Z-disk disorganization and protein aggregates' accumulation. Protein aggregates that do not fit the proteasome's narrow pore are targeted for removal via a specialized form of autophagy, called aggrephagy. Our study aims to investigate the potential pathogenic role of aggrephagy in 52 muscle samples from an Italian MFM multicentric cohort. We measured, the percentage of positive areas of key aggrephagy proteins by immunofluorescence staining, of sequestosome 1 (p62/SQSTM1), Neighbor of BRCA1 Gene 1 (NBR1), and ubiquitinated proteins (FK2) in 11 DES-, 6 DNAJB6-, 5 FLNC-, 18 MYOT- and 12 TTN-mutated patients. We showed that all aggrephagy markers are increased in these patients, regardless of the mutated genes, suggesting a possible common pathomechanism; no positive signal was found in healthy, age-matched controls. We analyzed the association between positivity levels of these markers, measured as percentage of positive areas, and selected clinical features utilizing generalized linear mixed models with gamma distribution as the probability model and center-specific random effects to better capture possible heterogeneity across participating centers. Our findings indicate significant associations between levels of p62, NBR1, and FK2 with age at biopsy (p62 and NBR1 p-values < 0.001, FK2 p-value < 0.05), age of onset (p62 and NBR1 p-values < 0.001, FK2 p-value < 0.01) and disease severity through Walton & Gardner-Medwin (WGM) score at biopsy (all p-values < 0.001) and at the last visit (all p-values < 0.05). Noteworthy, the aggrephagic pathway is mostly activated in MYOT-mutated patients compared to the other subgroups. Moreover, the association between aggrephagy and WGM score at biopsy is stronger in this subgroup. Overall, our study emphasizes the role of aggrephagy in MFMs across all patients, and its association with specific clinical parameters.
Keywords: Clinical association; Genetic rare diseases; Myofibrillar alterations; Protein aggregation.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: This study was approved by the Institutional Review Board at the Fondazione IRCCS Istituto Neurologico Carlo Besta (Project identification code 84/2022) in compliance with the current version of the Declaration of Helsinki as well as all national legal and regulatory requirements. Due to the observational, retrospective nature of the study and since the study is based on already available anonymous data, a written informed consent procedure is not needed. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures

References
-
- Palmio J, Udd B (2016) Myofibrillar and distal myopathies. Rev Neurol (Paris) 172(10):587–593. 10.1016/j.neurol.2016.07.019 - PubMed
-
- Bortolani S, Savarese M, Vattemi G et al (2024) Clinical, histopathologic, and genetic features of patients with myofibrillary and distal myopathies: experience from the Italian network. Neurology 103(4):e209697. 10.1212/WNL.0000000000209697 - PubMed
-
- Fichna JP, Maruszak A, Żekanowski C (2018) Myofibrillar myopathy in the genomic context. J Appl Genet 59(4):431–439. 10.1007/s13353-018-0463-4 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Miscellaneous
