

Mitochondria: the hidden engines of traumatic brain injury-driven neurodegeneration
- PMID: 40417416
- PMCID: PMC12098645
- DOI: 10.3389/fncel.2025.1570596
Mitochondria: the hidden engines of traumatic brain injury-driven neurodegeneration
Abstract
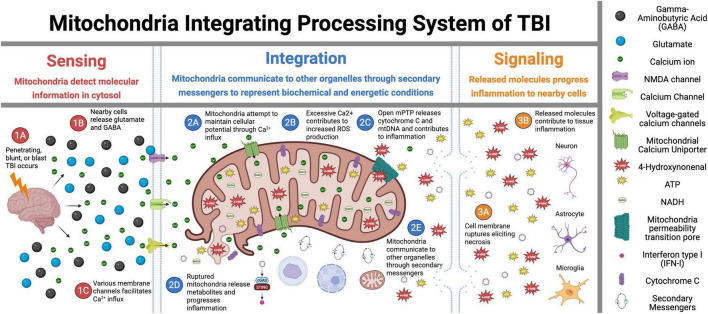
Mitochondria play a critical role in brain energy metabolism, cellular signaling, and homeostasis, making their dysfunction a key driver of secondary injury progression in traumatic brain injury (TBI). This review explores the relationship between mitochondrial bioenergetics, metabolism, oxidative stress, and neuroinflammation in the post-TBI brain. Mitochondrial dysfunction disrupts adenosine triphosphate (ATP) production, exacerbates calcium dysregulation, and generates reactive oxygen species, triggering a cascade of neuronal damage and neurodegenerative processes. Moreover, damaged mitochondria release damage-associated molecular patterns (DAMPs) such as mitochondrial DNA (mtDNA), Cytochrome C, and ATP, triggering inflammatory pathways that amplify tissue injury. We discuss the metabolic shifts that occur post-TBI, including the transition from oxidative phosphorylation to glycolysis and the consequences of metabolic inflexibility. Potential therapeutic interventions targeting mitochondrial dynamics, bioenergetic support, and inflammation modulation are explored, highlighting emerging strategies such as mitochondrial-targeted antioxidants, metabolic substrate supplementation, and pharmacological regulators of mitochondrial permeability transition pores. Understanding these mechanisms is crucial for developing novel therapeutic approaches to mitigate neurodegeneration and enhance recovery following brain trauma.
Keywords: bioenergetics; brain injury; metabolism; mitochondria; neurodegeneration.
Copyright © 2025 Olatona, Sterben, Kansakar, Symes and Liaudanskaya.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures





References
-
- Anderson R., Barger J., Edwards M., Braun K., O’Connor C., Prolla T., et al. (2008). Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell. 7 101–111. 10.1111/j.1474-9726.2007.00357.x - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources