

Spike-in enhanced phosphoproteomics uncovers synergistic signaling responses to MEK inhibition in colon cancer cells
- PMID: 40419504
- PMCID: PMC12106795
- DOI: 10.1038/s41467-025-59404-y
Spike-in enhanced phosphoproteomics uncovers synergistic signaling responses to MEK inhibition in colon cancer cells
Abstract
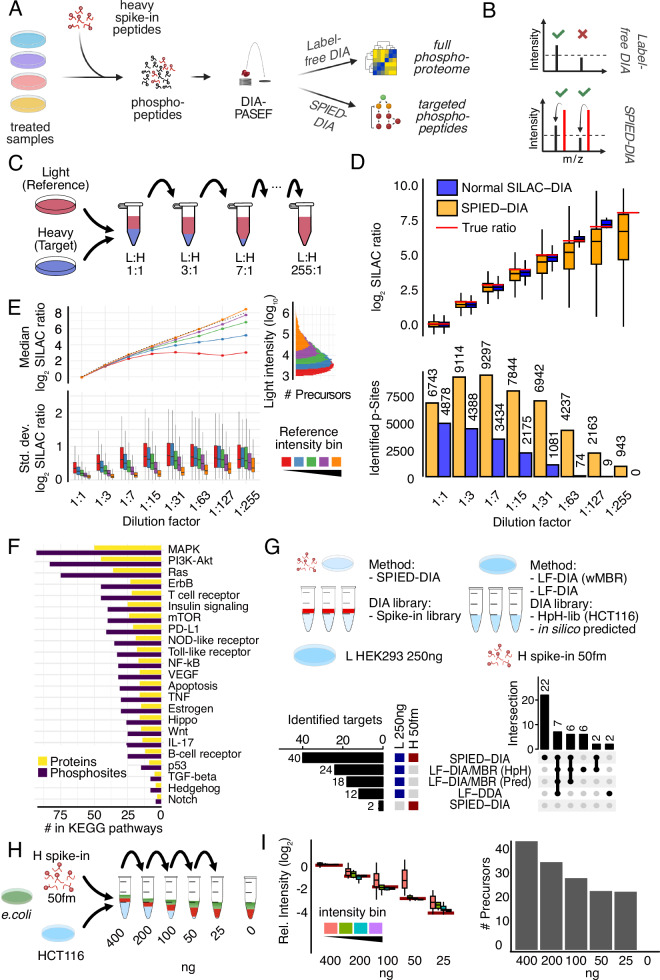
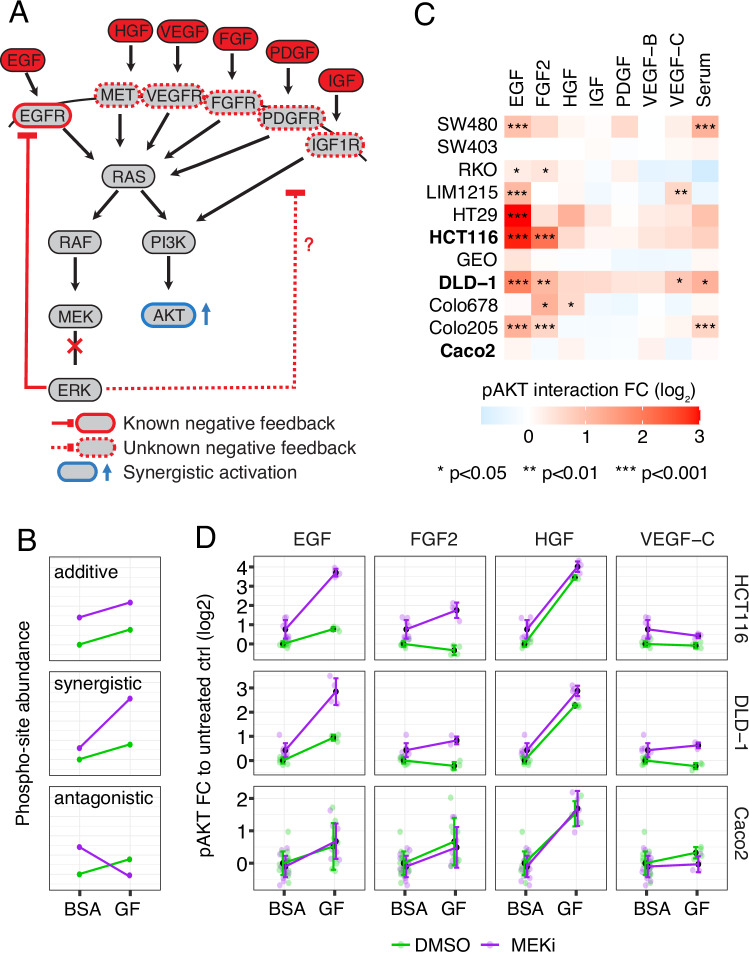
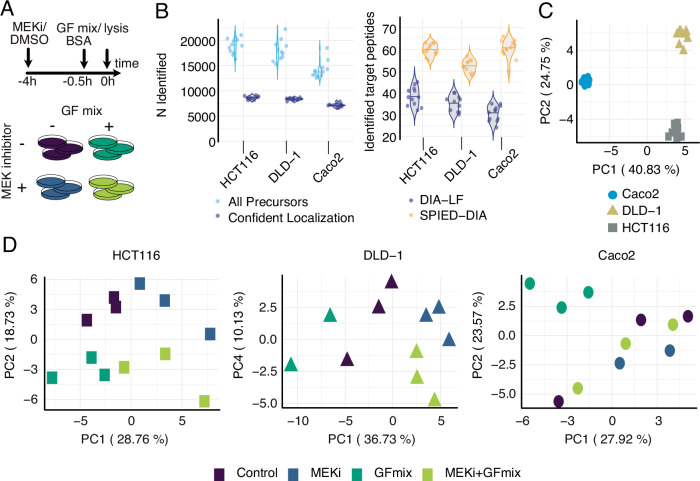
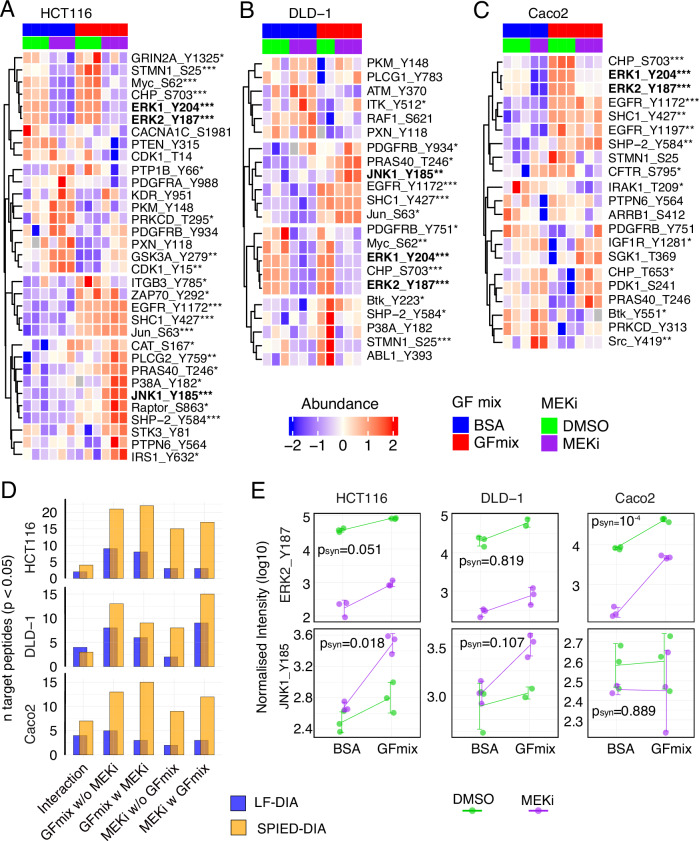
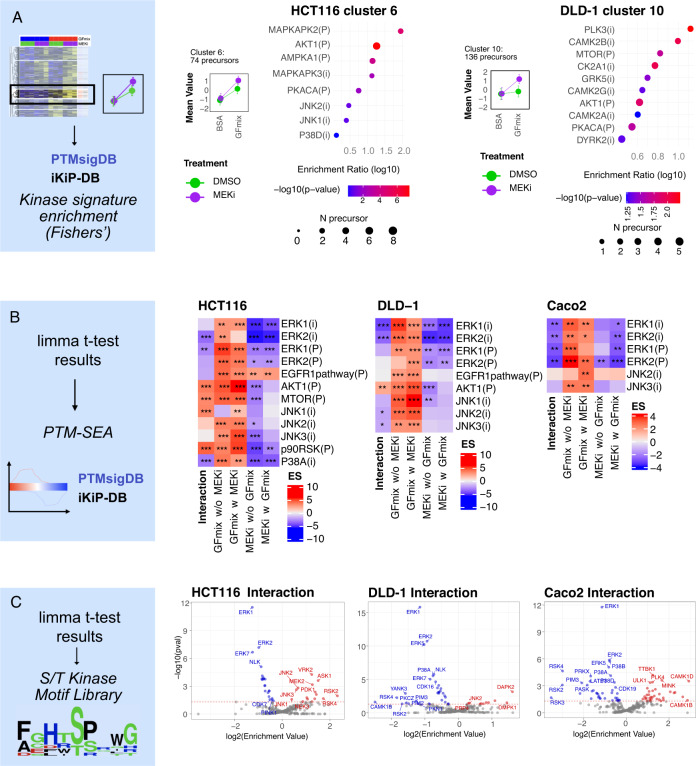
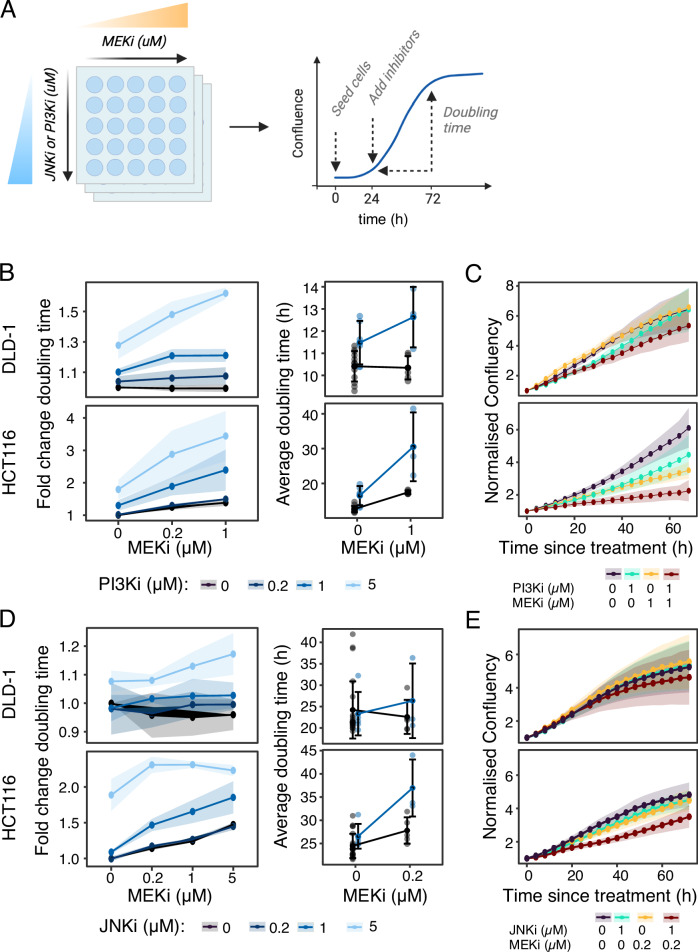
Targeted kinase inhibitors are a cornerstone of cancer therapy, but their success is often hindered by the complexity of cellular signaling networks that can lead to resistance. Overcoming this challenge necessitates a deep understanding of cellular signaling responses. While standard global phosphoproteomics offers extensive insights, lengthy processing times, the complexity of data interpretation, and frequent omission of crucial phosphorylation sites limit its utility. Here, we combine data-independent acquisition (DIA) with spike-in of synthetic heavy stable isotope-labeled phosphopeptides to facilitate the targeted detection of particularly informative phosphorylation sites. Our spike-in enhanced detection in DIA (SPIED-DIA) approach integrates the improved sensitivity of spike-in-based targeted detection with the discovery potential of global phosphoproteomics into a simple workflow. We employed this method to investigate synergistic signaling responses in colorectal cancer cell lines following MEK inhibition. Our findings highlight that combining MEK inhibition with growth factor stimulation synergistically activates JNK signaling in HCT116 cells. This synergy emphasizes the therapeutic potential of concurrently targeting MEK and JNK pathways, as evidenced by the significantly impaired growth of HCT116 cells when treated with both inhibitors. Our results demonstrate that SPIED-DIA effectively identifies synergistic signaling responses in colorectal cancer cells, presenting a valuable tool for uncovering new therapeutic targets and strategies in cancer treatment.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Effects of MEK inhibitors GSK1120212 and PD0325901 in vivo using 10-plex quantitative proteomics and phosphoproteomics.Proteomics. 2015 Jan;15(2-3):462-73. doi: 10.1002/pmic.201400154. Epub 2014 Oct 18. Proteomics. 2015. PMID: 25195567 Free PMC article.
-
Phosphoproteome analysis of the MAPK pathway reveals previously undetected feedback mechanisms.Proteomics. 2016 Jul;16(14):1998-2004. doi: 10.1002/pmic.201600119. Proteomics. 2016. PMID: 27273156
-
Phosphoproteomic analysis of leukemia cells under basal and drug-treated conditions identifies markers of kinase pathway activation and mechanisms of resistance.Mol Cell Proteomics. 2012 Aug;11(8):453-66. doi: 10.1074/mcp.M112.017483. Epub 2012 Apr 29. Mol Cell Proteomics. 2012. PMID: 22547687 Free PMC article.
-
Dual Inhibition of MEK and PI3K Pathway in KRAS and BRAF Mutated Colorectal Cancers.Int J Mol Sci. 2015 Sep 23;16(9):22976-88. doi: 10.3390/ijms160922976. Int J Mol Sci. 2015. PMID: 26404261 Free PMC article. Review.
-
Biomarker assays for phosphorylated MAP kinase. Their utility for measurement of MEK inhibition.Methods Mol Med. 2003;85:31-8. doi: 10.1385/1-59259-380-1:31. Methods Mol Med. 2003. PMID: 12710194 Review. No abstract available.
Cited by
-
Single-nucleus proteomics identifies regulators of protein transport.bioRxiv [Preprint]. 2024 Jun 18:2024.06.17.599449. doi: 10.1101/2024.06.17.599449. bioRxiv. 2024. PMID: 38948785 Free PMC article. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
