

Cellular hierarchies of embryonal tumors with multilayered rosettes are shaped by oncogenic microRNAs and receptor-ligand interactions
- PMID: 40419763
- PMCID: PMC12202505
- DOI: 10.1038/s43018-025-00964-9
Cellular hierarchies of embryonal tumors with multilayered rosettes are shaped by oncogenic microRNAs and receptor-ligand interactions
Abstract
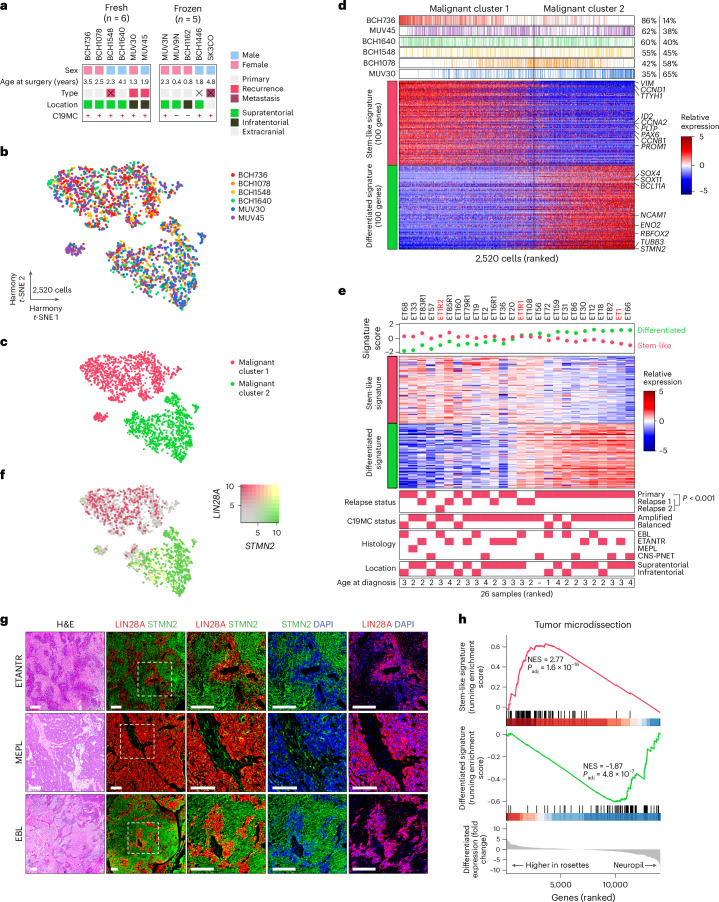
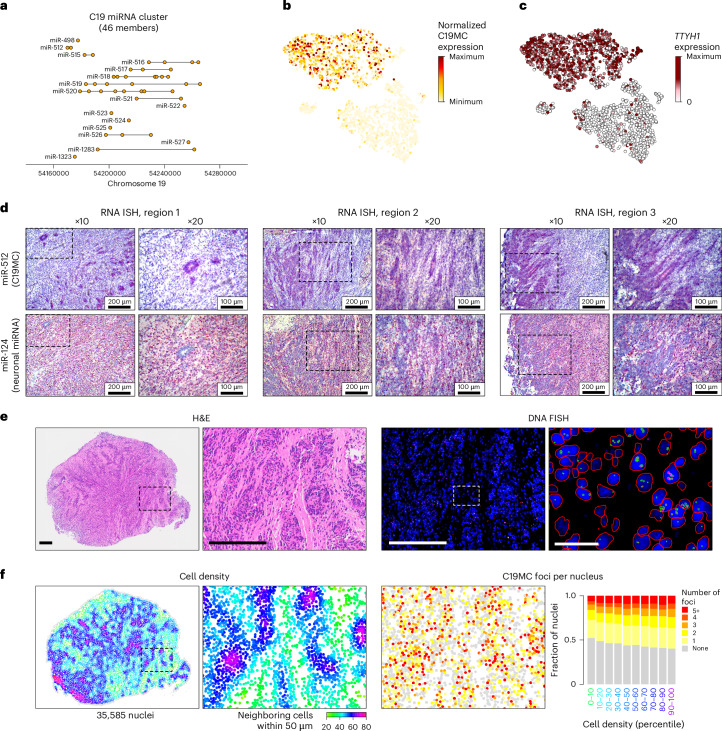
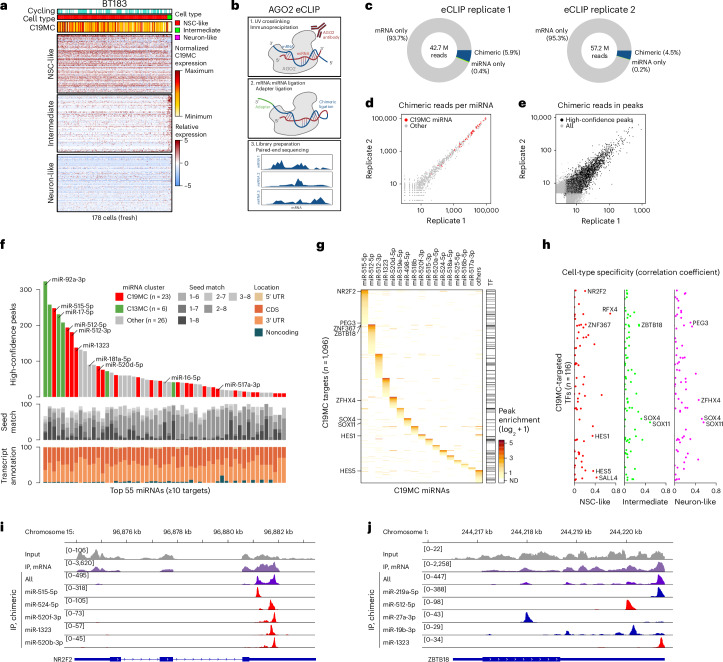
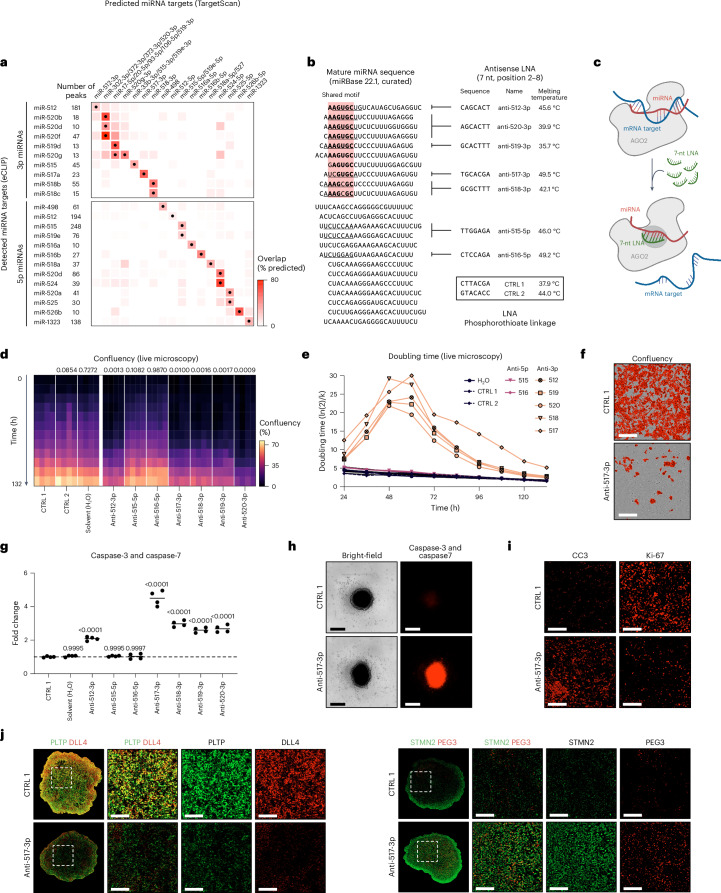
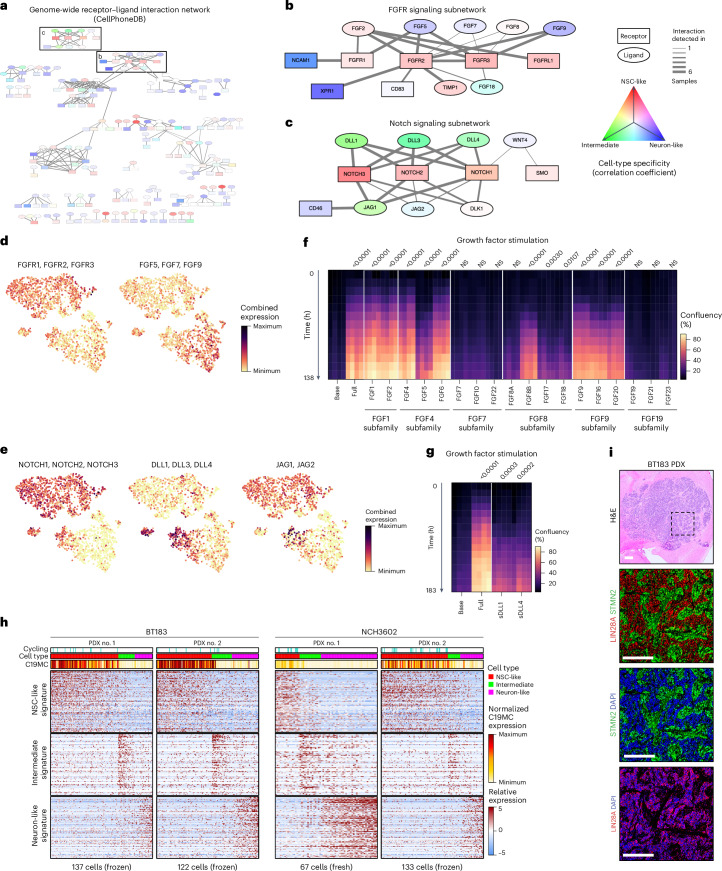
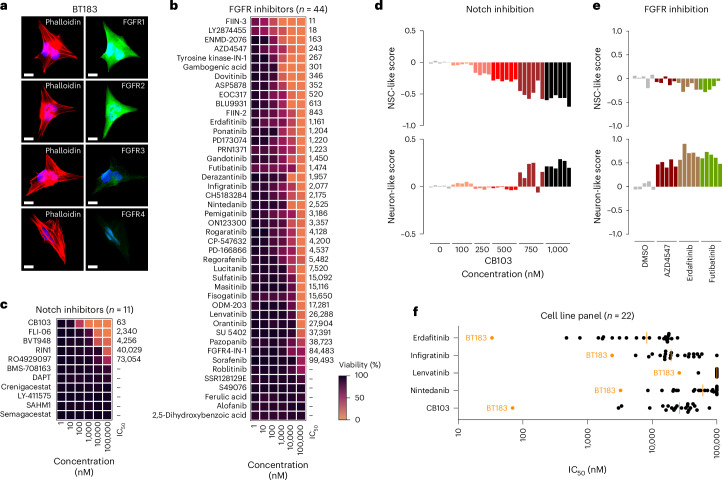
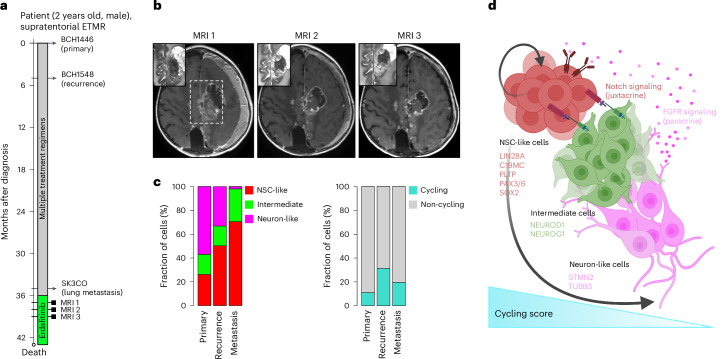
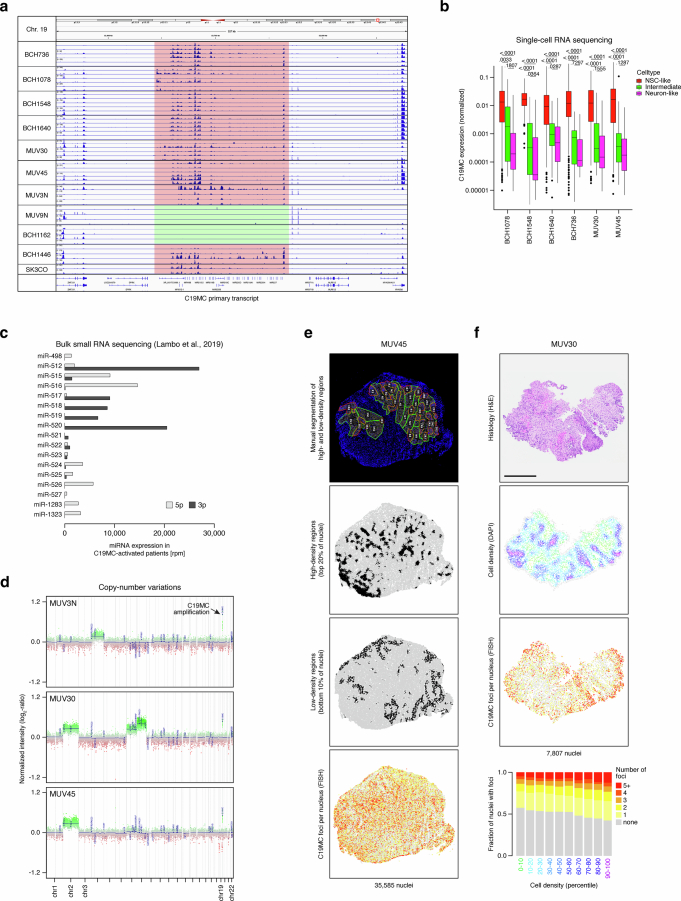
Embryonal tumor with multilayered rosettes (ETMR) is a pediatric brain tumor with dismal prognosis. Characteristic alterations of the chromosome 19 microRNA cluster (C19MC) are observed in most ETMR; however, the ramifications of C19MC activation and the complex cellular architecture of ETMR remain understudied. Here we analyze 11 ETMR samples from patients using single-cell transcriptomics and multiplexed spatial imaging. We reveal a spatially distinct cellular hierarchy that spans highly proliferative neural stem-like cells and more differentiated neuron-like cells. C19MC is predominantly expressed in stem-like cells and controls a transcriptional network governing stemness and lineage commitment, as resolved by genome-wide analysis of microRNA-mRNA binding. Systematic analysis of receptor-ligand interactions between malignant cell types reveals fibroblast growth factor receptor and Notch signaling as oncogenic pathways that can be successfully targeted in preclinical models and in one patient with ETMR. Our study provides fundamental insights into ETMR pathobiology and a powerful rationale for more effective targeted therapies.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: M.G.F. is a consultant for Twentyeight-Seven Therapeutics and Blueprint Medicines. J.C.T. serves as a consultant for Advanced Accelerator Applications-Novartis. The other authors declare no competing interests.
Figures










References
-
- Khan, S. et al. Clinical phenotypes and prognostic features of embryonal tumours with multi-layered rosettes: a Rare Brain Tumor Registry study. Lancet Child Adolesc. Health5, 800–813 (2021). - PubMed
-
- Neumann, J. E. et al. A mouse model for embryonal tumors with multilayered rosettes uncovers the therapeutic potential of Sonic-hedgehog inhibitors. Nat. Med.23, 1191–1202 (2017). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
