

DRaCOon: a novel algorithm for pathway-level differential co-expression analysis in transcriptomics
- PMID: 40419963
- PMCID: PMC12107744
- DOI: 10.1186/s12859-025-06162-9
DRaCOon: a novel algorithm for pathway-level differential co-expression analysis in transcriptomics
Abstract
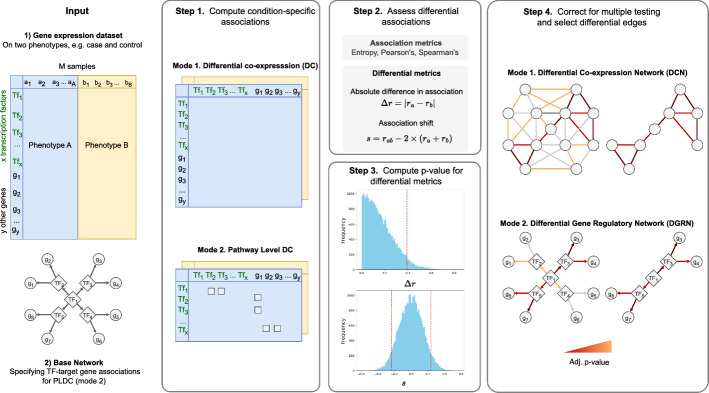
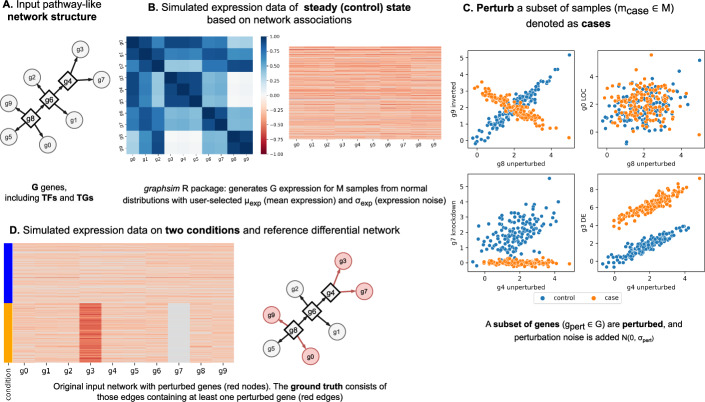
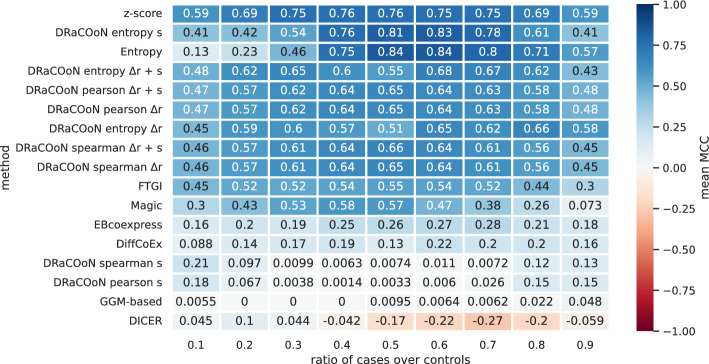
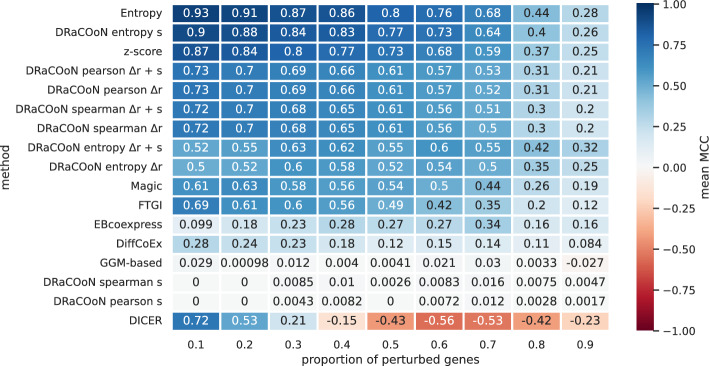
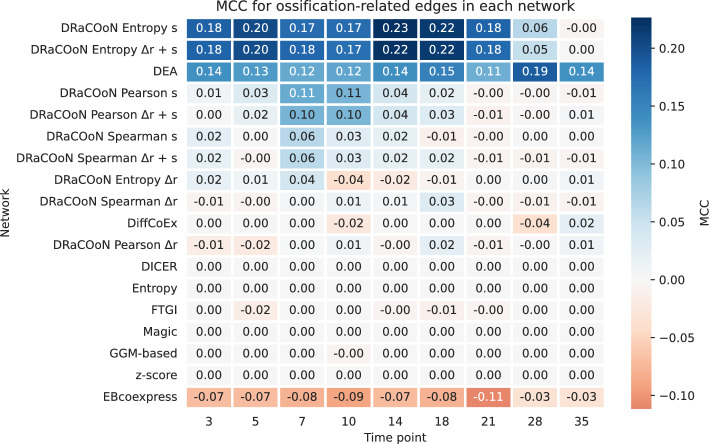
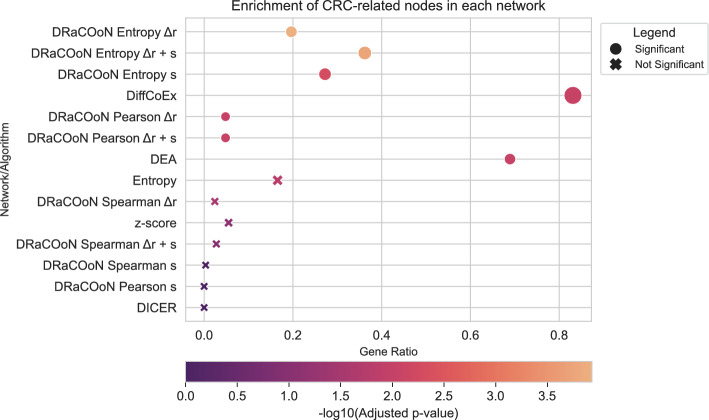
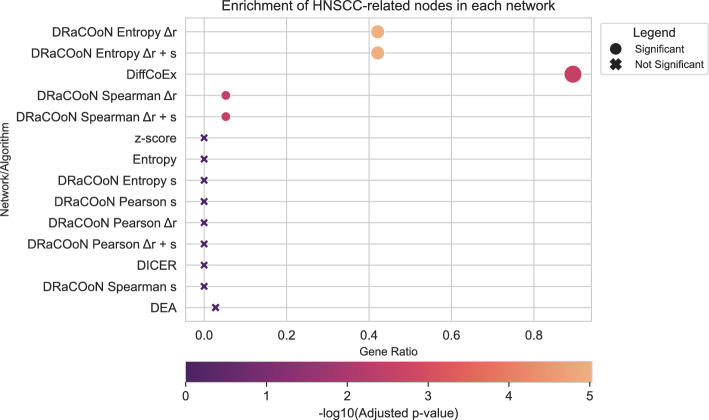
Understanding the molecular mechanisms underlying diseases is crucial for more precise, personalized medicine. Pathway-level differential co-expression analysis, a powerful approach for transcriptomics, identifies condition-specific changes in gene-gene interaction networks, offering targeted insights. However, a key challenge is the lack of robust methods and benchmarks specifically for evaluating algorithms' ability to identify disrupted gene-gene associations across conditions. We introduce DRaCOoN (Differential Regulatory and Co-expression Networks), a Python package and web tool for pathway-level differential co-expression analysis. DRaCOoN uniquely integrates multiple association and differential metrics, with a novel, computationally efficient permutation test for significance assessment. Crucially, DRaCOoN also provides a benchmarking framework for comprehensive method evaluation. Extensive benchmarking on simulated data and three real-world datasets (bone healing, colorectal cancer, and head/neck carcinoma) showed that DRaCOoN, particularly with an entropy-based association measure and the s differential metric, consistently outperforms eight other methods. It remains highly accurate in balanced datasets, robust to varying gene perturbation levels, and identifies biologically relevant regulatory changes. Furthermore, DRaCOoN serves as both a powerful tool and a benchmarking framework for elucidating disease mechanisms from transcriptomics data, advancing precision medicine by uncovering critical gene regulatory alterations.
Keywords: Differential networking; Differential regulation; Disease module identification; Network-based gene expression analysis; Pathway-level differential co-expression.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare no Conflict of interest. Authors’ information: Not applicable.
Figures

Similar articles
-
GSNCASCR: An R Package to Identify Differentially Co-Expressed Curated Gene Sets with Single-Cell RNA-Seq Data.Int J Mol Sci. 2025 May 16;26(10):4771. doi: 10.3390/ijms26104771. Int J Mol Sci. 2025. PMID: 40429912 Free PMC article.
-
Benchmarking algorithms for spatially variable gene identification in spatial transcriptomics.Bioinformatics. 2025 Mar 29;41(4):btaf131. doi: 10.1093/bioinformatics/btaf131. Bioinformatics. 2025. PMID: 40139667 Free PMC article.
-
Identification of regulatory modules in genome scale transcription regulatory networks.BMC Syst Biol. 2017 Dec 15;11(1):140. doi: 10.1186/s12918-017-0493-2. BMC Syst Biol. 2017. PMID: 29246163 Free PMC article.
-
Learning Differential Module Networks Across Multiple Experimental Conditions.Methods Mol Biol. 2019;1883:303-321. doi: 10.1007/978-1-4939-8882-2_13. Methods Mol Biol. 2019. PMID: 30547406 Review.
-
Differential Co-Expression Analyses Allow the Identification of Critical Signalling Pathways Altered during Tumour Transformation and Progression.Int J Mol Sci. 2020 Dec 12;21(24):9461. doi: 10.3390/ijms21249461. Int J Mol Sci. 2020. PMID: 33322692 Free PMC article. Review.
References
-
- Chowdhury HA, Bhattacharyya DK, Kalita JK. (Differential) co-expression analysis of gene expression: a survey of best practices. IEEE/ACM Trans Comput Biol Bioinform. 2020;17(4):1154–73. 10.1109/TCBB.2019.2893170. - PubMed
-
- Kakati T, Bhattacharyya DK, Barah P, Kalita JK. Comparison of methods for differential co-expression analysis for disease biomarker prediction. Comput Biol Med. 2019;113: 103380. 10.1016/j.compbiomed.2019.103380. - PubMed
-
- Mirlekar B. Co-expression of master transcription factors determines cd4+ t cell plasticity and functions in auto-inflammatory diseases. Immunol Lett. 2020;222:58–66. 10.1016/j.imlet.2020.03.007. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
