

A case of congenital heart defects and familial exudative vitreoretinopathy caused by activation of a cryptic splice donor in NOTCH1
- PMID: 40420130
- PMCID: PMC12105292
- DOI: 10.1186/s12920-025-02160-1
A case of congenital heart defects and familial exudative vitreoretinopathy caused by activation of a cryptic splice donor in NOTCH1
Abstract
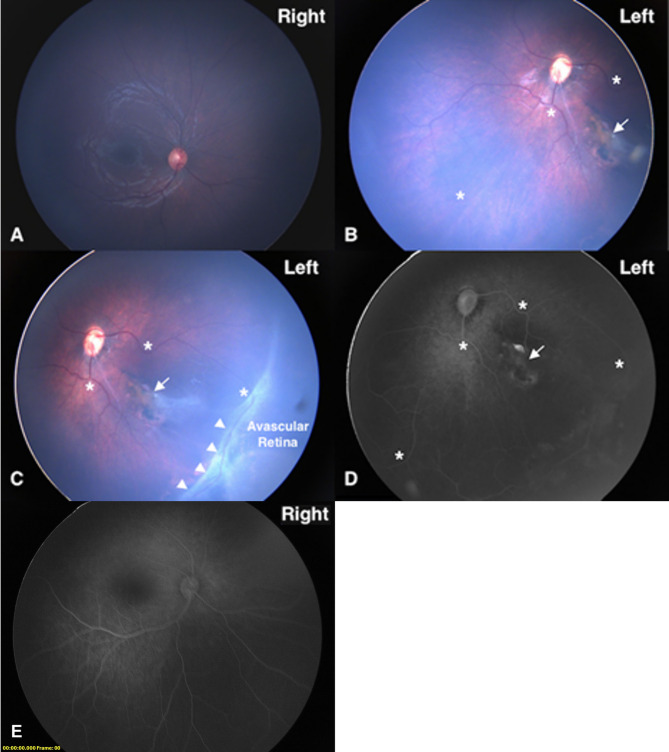
Background: NOTCH1 is associated with two disorders of vascular development, Adams-Oliver Syndrome 5 (AOS5) and aortic valve disease 1 (AOVD1). Here we report a disease-causing variant in NOTCH1 that has a previously undemonstrated effect on splicing. Additionally, we found that the proband has the optic phenotype of familial exudative vitreoretinopathy (FEVR) which has been reported for probands with pathogenic variants in genes in the notch signaling pathway, but never for NOTCH1.
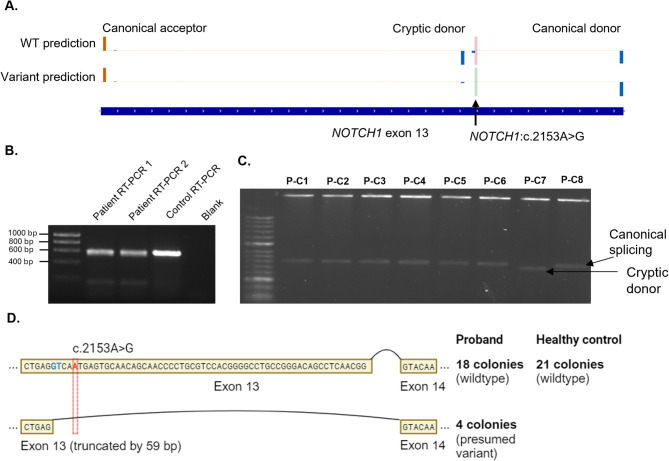
Case presentation: The proband presented with a ventricular septal defect, pulmonic stenosis, and ocular findings consistent with familial exudative vitreoretinopathy (FEVR), which NOTCH1 has not been associated with to date. Trio exome sequencing identified a paternally inherited variant of uncertain significance in NOTCH1:c.2153 A > G. We assessed the variant's effect using RT-PCR, finding an increased use of a cryptic donor compared to the control. On this basis, we were able to re-classify this variant as pathogenic.
Conclusions: We expand the phenotypic spectrum of NOTCH1 and contribute to the building evidence that variants in NOTCH1 cause a spectrum of disorders of vascular development.
Keywords: NOTCH1; Adams-oliver syndrome; Familial exudative vitreoretinopathy; Rare disease; Splice variant.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: This study was approved under Mayo Clinic Institutional Review Board #19-003389. Informed consent to participate was obtained from all of the participants in the study. Informed consent to participate was obtained from the parents or legal guardians of any participant under the age of 16. The study adhered to the Declaration of Helsinki. Consent for publication: Written informed consent for publication of clinical details and/or clinical images was obtained from the parents of the patient. Competing interests: The authors declare no competing interests.
Figures

Similar articles
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
-
Trio-WES and functional validation reveals a novel splice site variant of SLC26A3 in a case with congenital chloride diarrhea and a systematic review of SLC26A3 mutations in China.Mol Genet Genomics. 2025 Mar 6;300(1):28. doi: 10.1007/s00438-025-02233-x. Mol Genet Genomics. 2025. PMID: 40047934
-
Familial Exudative Vitreoretinopathy-Like Retinal Findings in Adams-Oliver Syndrome Type 2.Clin Exp Ophthalmol. 2025 Jul 30. doi: 10.1111/ceo.14594. Online ahead of print. Clin Exp Ophthalmol. 2025. PMID: 40734490
-
Individual-level interventions to reduce personal exposure to outdoor air pollution and their effects on people with long-term respiratory conditions.Cochrane Database Syst Rev. 2021 Aug 9;8(8):CD013441. doi: 10.1002/14651858.CD013441.pub2. Cochrane Database Syst Rev. 2021. PMID: 34368949 Free PMC article.
-
Antipsychotics for schizophrenia spectrum disorders with catatonic symptoms.Cochrane Database Syst Rev. 2022 Jul 12;7(7):CD013100. doi: 10.1002/14651858.CD013100.pub2. Cochrane Database Syst Rev. 2022. PMID: 35844143 Free PMC article.
References
-
- Hassed S, Li S, Mulvihill J, Aston C, Palmer S. Adams-Oliver syndrome review of the literature: refining the diagnostic phenotype. Am J Med Genet A. 2017;173(3):790–800. 10.1002/ajmg.a.37889 - PubMed
-
- Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437(7056):270–4. 10.1038/nature03940 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
